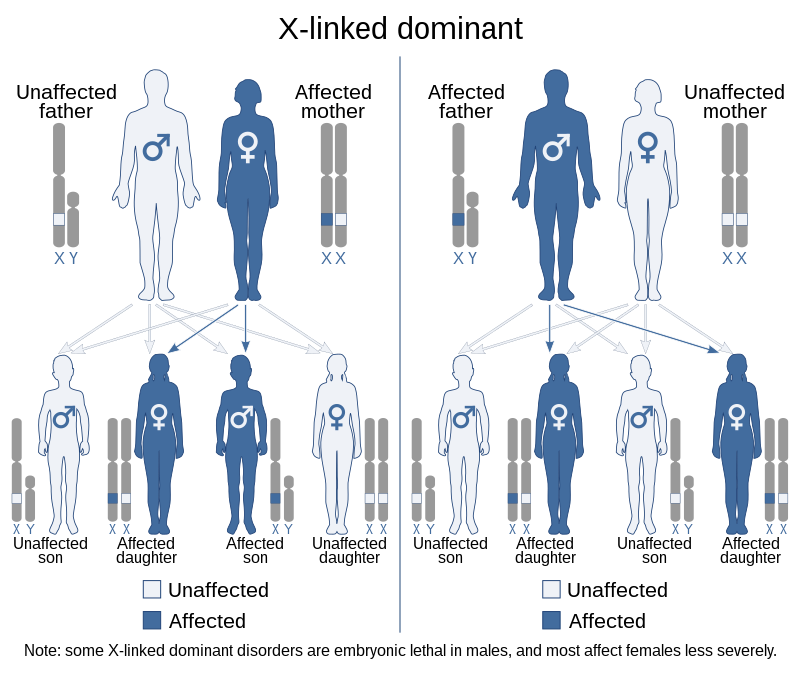
X-linked dominant inheritance
X-linked dominant inheritance, sometimes referred to as X-linked dominance, is a mode of genetic inheritance by which a dominant gene is carried on the X chromosome. As an inheritance pattern, it is less common than the X-linked recessive type. In medicine, X-linked dominant inheritance indicates that a gene responsible for a genetic disorder is located on the X chromosome, and only one copy of the allele is sufficient to cause the disorder when inherited from a parent who has the disorder. In this case, someone who expresses an X-linked dominant allele will exhibit the disorder and be considered affected.
X-linked dominant traits do not necessarily affect males more than females (unlike X-linked recessive traits). The exact pattern of inheritance varies, depending on whether the father or the mother has the trait of interest. All fathers that are affected by an X-linked dominant disorder will have affected daughters but not affected sons. However, if the mother is also affected then sons will have a chance of being affected, depending on whether a dominant or recessive X chromosome is passed on. When the son is affected, the mother will always be affected. Some X-linked dominant conditions are embryonic lethal in males, making them appear to only occur in females.[citation needed]
Genetics
As the X chromosome is one of the sex chromosomes (the other being the Y chromosome), X-linked inheritance is determined by the sex of the parent carrying a specific gene and can often seem complex. This is due to the fact that, typically, females have two copies of the X-chromosome, while males have only one copy. The difference between dominant and recessive inheritance patterns also plays a role in determining the chances of a child inheriting an X-linked disorder from their parentage.[citation needed]
Males can only get an X chromosome from their mother whilst females get an X chromosome from both parents. As a result, females tend to show higher prevalence of X-linked dominant disorders because they have more of a chance to inherit a faulty X chromosome.[citation needed]
Inheritance
In X-linked dominant inheritance, when the mother alone is the carrier of a mutated, or defective gene associated with a disease or disorder; she herself will have the disorder. Her children will inherit the disorder as follows:
- Of her daughters and sons: 50% will have the disorder, 50% will be completely unaffected. Children of either sex have an even chance of receiving either of their mother’s two X chromosomes, one of which contains the defective gene in question.
When the father alone is the carrier of a defective gene associated with a disease or disorder, he too will have the disorder. His children will inherit the disorder as follows:
- Of his daughters: 100% will have the disorder, since all of his daughters will receive one copy of his single X chromosome.
- Of his sons: none will have the disorder; sons do not receive an X chromosome from their father.
If both parents were carriers of a defective gene associated with a disease or disorder, they would both have the disorder. Their children would inherit the disorder as follows:
- Of their daughters: 100% will have the disorder, since all of the daughters will receive a copy of their father’s X chromosome.
- Of the sons: 50% will have the disorder, 50% will be completely unaffected. Sons have an equal chance of receiving either of their mother’s X chromosomes.
In such a case, where both parents carry and thus are affected by an X-linked dominant disorder, the chance of a daughter receiving two copies of the X chromosome with the defective gene is 50%, since daughters receive one copy of the X chromosome from both parents. Were this to occur with an X-linked dominant disorder, that daughter would likely experience a more severe form.
Some X-linked dominant conditions such as Aicardi syndrome are fatal to boys; therefore only girls with these conditions survive, or boys with Klinefelter’s syndrome (and hence have more than one X chromosome).
A few scholars have suggested discontinuing the use of the terms dominant and recessive when referring to X-linked inheritance, stating that the highly variable penetrance of X-linked traits in females as a result of mechanisms such as skewed X-inactivation or somatic mosaicism is difficult to reconcile with standard definitions of dominance and recessiveness.[1]
List of dominant X-linked diseases
- Vitamin D resistant rickets: X-linked hypophosphatemia
- Rett syndrome (95% of cases are due to sporadic mutations)
- Most cases of Alport syndrome[2]
- Incontinentia pigmenti[3][4]
- Giuffrè–Tsukahara syndrome[5]
- Goltz syndrome
- X-linked dominant porphyria[6]
- Aicardi Syndrome
See also
References
^ Dobyns WB, Filauro A, Tomson BN, Chan AS, Ho AW, Ting NT, Oosterwijk JC, Ober C (August 2004). “Inheritance of most X-linked traits is not dominant or recessive, just X-linked”. American Journal of Medical Genetics. Part A. 129A (2): 136–43. doi:10.1002/ajmg.a.30123. PMID 15316978.
^ Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. (October 2003). “X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study”. Journal of the American Society of Nephrology. 14 (10): 2603–10. doi:10.1097/01.ASN.0000090034.71205.74. PMID 14514738.
^ Ngan V (2005). “Incontinentia pigmenti”. DermNet NZ.
^ Incontinentia Pigmenti at eMedicine
^ Dalal AB, Sarkar A, Priya TP, Nandineni MR (August 2010). “Giuffrè-Tsukahara syndrome: Evidence for X-linked dominant inheritance and review”. American Journal of Medical Genetics. Part A. 152A (8): 2057–60. doi:10.1002/ajmg.a.33505. PMID 20635354.
^ Seager MJ, Whatley SD, Anstey AV, Millard TP (January 2014). “X-linked dominant protoporphyria: a new porphyria”. Clinical and Experimental Dermatology. 39 (1): 35–7. doi:10.1111/ced.12202. PMID 24131146.
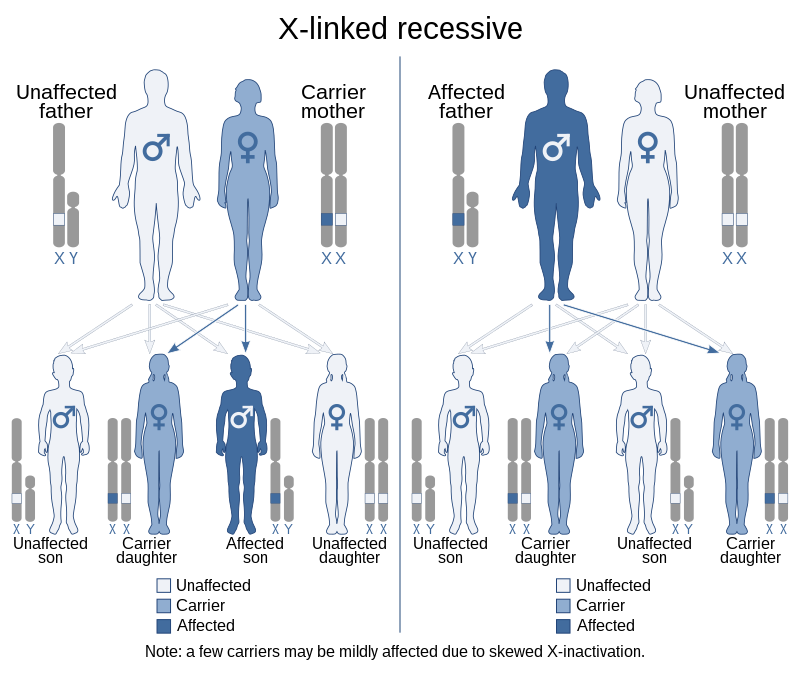
X-linked recessive inheritance
X-linked recessive inheritance is a mode of inheritance in which a mutation in a gene on the X chromosome causes the phenotype to be always expressed in males (who are necessarily homozygous for the gene mutation because they have one X and one Y chromosome) and in females who are homozygous for the gene mutation, see zygosity. Females with one copy of the mutated gene are carriers.[citation needed]
X-linked inheritance means that the gene causing the trait or the disorder is located on the X chromosome. Females have two X chromosomes while males have one X and one Y chromosome. Carrier females who have only one copy of the mutation do not usually express the phenotype, although differences in X-chromosome inactivation (known as skewed X-inactivation) can lead to varying degrees of clinical expression in carrier females, since some cells will express one X allele and some will express the other. The current estimate of sequenced X-linked genes is 499, and the total, including vaguely defined traits, is 983.[1]
Patterns of inheritance
{kind=link}
In humans, inheritance of X-linked recessive traits follows a unique pattern made up of three points.
- The first is that affected fathers cannot pass X-linked recessive traits to their sons because fathers give Y chromosomes to their sons. This means that males affected by an X-linked recessive disorder inherited the responsible X chromosome from their mothers.
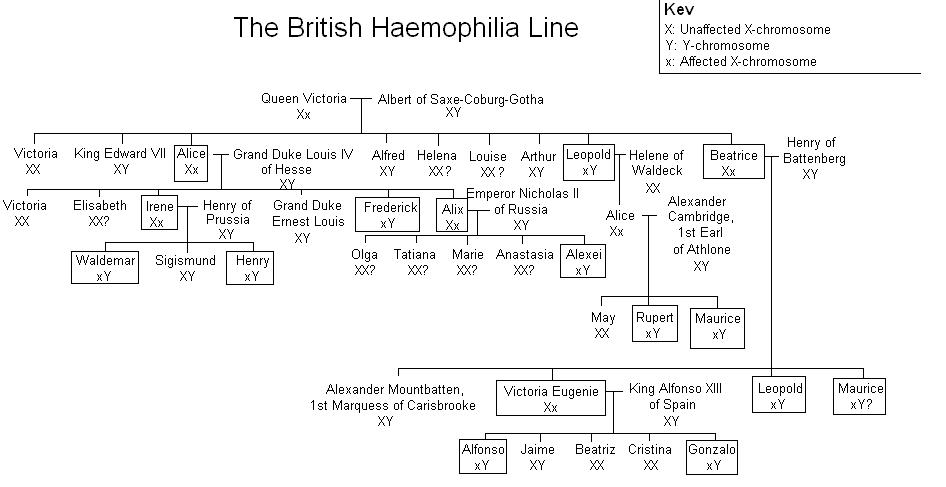
- Second, X-linked recessive traits are more commonly expressed in males than females.[2] This is due to the fact that males possess only a single X chromosome, and therefore require only one mutated X in order to be affected. Women possess two X chromosomes, and thus must receive two of the mutated recessive X chromosomes (one from each parent). A popular example showing this pattern of inheritance is that of the descendants of Queen Victoria and the blood disease hemophilia.[3]
- The last pattern seen is that X-linked recessive traits tend to skip generations, meaning that an affected grandfather will not have an affected son, but could have an affected grandson through his daughter.[4] Explained further, all daughters of an affected man will obtain his mutated X, and will then be either carriers or affected themselves depending on the mother. The resulting sons will either have a 50% chance of being affected (mother is carrier), or 100% chance (mother is affected). It is because of these percentages that we see males more commonly affected than females.
Pushback on recessive/dominant terminology
A few scholars have suggested discontinuing the use of the terms dominant and recessive when referring to X-linked inheritance.[5] The possession of two X chromosomes in females leads to dosage issues which are alleviated by X-inactivation.[6] Stating that the highly variable penetrance of X-linked traits in females as a result of mechanisms such as skewed X-inactivation or somatic mosaicism is difficult to reconcile with standard definitions of dominance and recessiveness, scholars have suggested referring to traits on the X chromosome simply as X-linked.[5]
Examples
Most common
The most common X-linked recessive disorders are:[7]
- Red–green color blindness, a very common trait in humans and frequently used to explain X-linked disorders.[8] Between seven and ten percent of men and 0.49% to 1% of women are affected. Its commonness may be explained by its relatively benign nature. It is also known as daltonism.
- Hemophilia A, a blood clotting disorder caused by a mutation of the Factor VIII gene and leading to a deficiency of Factor VIII. It was once thought to be the “royal disease” found in the descendants of Queen Victoria. This is now known to have been Hemophilia B (see below).[9][10]
- Hemophilia B, also known as Christmas disease,[11] a blood clotting disorder caused by a mutation of the Factor IX gene and leading to a deficiency of Factor IX. It is rarer than hemophilia A. As noted above, it was common among the descendants of Queen Victoria.
- Duchenne muscular dystrophy, which is associated with mutations in the dystrophin gene. It is characterized by rapid progression of muscle degeneration, eventually leading to loss of skeletal muscle control, respiratory failure, and death.
- Becker’s muscular dystrophy, a milder form of Duchenne, which causes slowly progressive muscle weakness of the legs and pelvis.
- X-linked ichthyosis, a form of ichthyosis caused by a hereditary deficiency of the steroid sulfatase (STS) enzyme. It is fairly rare, affecting one in 2,000 to one in 6,000 males.[12]
- X-linked agammaglobulinemia (XLA), which affects the body’s ability to fight infection. XLA patients do not generate mature B cells.[13] B cells are part of the immune system and normally manufacture antibodies (also called immunoglobulins) which defends the body from infections (the humoral response). Patients with untreated XLA are prone to develop serious and even fatal infections.[14]
- Glucose-6-phosphate dehydrogenase deficiency, which causes nonimmune hemolytic anemia in response to a number of causes, most commonly infection or exposure to certain medications, chemicals, or foods. Commonly known as “favism”, as it can be triggered by chemicals existing naturally in broad (or fava) beans.[15]
Less common disorders
See also: X-linked intellectual disability
Theoretically, a mutation in any of the genes on chromosome X may cause disease, but below are some notable ones, with short description of symptoms:
- Adrenoleukodystrophy; leads to progressive brain damage, failure of the adrenal glands and eventually death.
- Alport syndrome; glomerulonephritis, endstage kidney disease, and hearing loss. A minority of Alport syndrome cases are due to an autosomal recessive mutation in the gene coding for type IV collagen.
- Androgen insensitivity syndrome; variable degrees of undervirilization and/or infertility in XY persons of either sex
- Barth syndrome; metabolism distortion, delayed motor skills, stamina deficiency, hypotonia, chronic fatigue, delayed growth, cardiomyopathy, and compromised immune system.
- Blue cone monochromacy; low vision acuity, color blindness, photophobia, infantile nystagmus.
- Centronuclear myopathy; where cell nuclei are abnormally located in skeletal muscle cells. In CNM the nuclei are located at a position in the center of the cell, instead of their normal location at the periphery.
- Charcot–Marie–Tooth disease (CMTX2-3); disorder of nerves (neuropathy) that is characterized by loss of muscle tissue and touch sensation, predominantly in the feet and legs but also in the hands and arms in the advanced stages of disease.
- Coffin–Lowry syndrome; severe intellectual disability sometimes associated with abnormalities of growth, cardiac abnormalities, kyphoscoliosis as well as auditory and visual abnormalities.
- Fabry disease; A lysosomal storage disease causing anhidrosis, fatigue, angiokeratomas, burning extremity pain and ocular involvement.
- Hunter syndrome; potentially causing hearing loss, thickening of the heart valves leading to a decline in cardiac function, obstructive airway disease, sleep apnea, and enlargement of the liver and spleen.
- Hypohidrotic ectodermal dysplasia, presenting with hypohidrosis, hypotrichosis, hypodontia
- Kabuki syndrome (the KDM6A variant); multiple congenital anomalies and intellectual disability.
- Spinal and bulbar muscular atrophy; muscle cramps and progressive weakness
- Lesch–Nyhan syndrome; neurologic dysfunction, cognitive and behavioral disturbances including self-mutilation, and uric acid overproduction (hyperuricemia)
- Lowe syndrome; hydrophthalmia, cataracts, intellectual disabilities, aminoaciduria, reduced renal ammonia production and vitamin D-resistant rickets
- Menkes disease; sparse and coarse hair, growth failure, and deterioration of the nervous system
- Nasodigitoacoustic syndrome; misshaped nose, brachydactyly of the distal phalanges, sensorineural deafness
- Nonsyndromic deafness; hearing loss
- Norrie disease; cataracts, leukocoria along with other developmental issues in the eye
- Occipital horn syndrome; deformations in the skeleton
- Ocular albinism; lack of pigmentation in the eye
- Ornithine transcarbamylase deficiency; developmental delay and intellectual disability. Progressive liver damage, skin lesions, and brittle hair may also be seen
- Oto-palato-digital syndrome; facial deformities, cleft palate, hearing loss
- Siderius X-linked mental retardation syndrome; cleft lip and palate with intellectual disability and facial dysmorphism, caused by mutations in the histone demethylase PHF8
- Simpson–Golabi–Behmel syndrome; coarse faces with protruding jaw and tongue, widened nasal bridge, and upturned nasal tip
- Spinal muscular atrophy caused by UBE1 gene mutation; weakness due to loss of the motor neurons of the spinal cord and brainstem
- Wiskott–Aldrich syndrome; eczema, thrombocytopenia, immune deficiency, and bloody diarrhea
- X-linked severe combined immunodeficiency (SCID); infections, usually causing death in the first years of life
- X-linked sideroblastic anemia; skin paleness, fatigue, dizziness and enlarged spleen and liver.
See also
References
- ^ “OMIM X-linked Genes”. nih.gov. Archived from the original on 7 March 2016. Retrieved 3 May 2018.
- ^ Understanding Genetics: A New York, Mid-Atlantic Guide for Patients and Health Professionals. National Center for Biotechnology Information. 8 July 2009. Retrieved 9 June 2020.
- ^ “History of Bleeding Disorders”. National Hemophilia Foundation. 2014-03-04. Retrieved 2020-06-09.
- ^ Pierce, Benjamin A. (2020). Genetics: A Conceptual Approach. Macmillan Learning. pp. 154–155. ISBN 978-1-319-29714-5.
- ^ Jump up to:a b Dobyns, William B.; Filauro, Allison; Tomson, Brett N.; Chan, April S.; Ho, Allen W.; Ting, Nicholas T.; Oosterwijk, Jan C.; Ober, Carole (2004). “Inheritance of most X-linked traits is not dominant or recessive, just X-linked”. American Journal of Medical Genetics. 129A (2): 136–43. doi:10.1002/ajmg.a.30123. PMID 15316978. S2CID 42108591.
- ^ Shvetsova, Ekaterina; Sofronova, Alina; Monajemi, Ramin; Gagalova, Kristina; Draisma, Harmen H. M.; White, Stefan J.; Santen, Gijs W. E.; Chuva de Sousa Lopes, Susana M.; Heijmans, Bastiaan T.; van Meurs, Joyce; Jansen, Rick (March 2019). “Skewed X-inactivation is common in the general female population”. European Journal of Human Genetics. 27 (3): 455–465. doi:10.1038/s41431-018-0291-3. ISSN 1476-5438. PMC 6460563. PMID 30552425.
- ^ GP Notebook – X-linked recessive disorders Archived 2011-06-13 at the Wayback Machine Retrieved on 5 Mars, 2009
- ^ “OMIM Color Blindness, Deutan Series; CBD”. nih.gov. Archived from the original on 29 September 2009. Retrieved 3 May 2018.
- ^ Michael Price (8 October 2009). “Case Closed: Famous Royals Suffered From Hemophilia”. ScienceNOW Daily News. AAAS. Archived from the original on 20 October 2013. Retrieved 9 October 2009.
- ^ Rogaev, Evgeny I.; Grigorenko, Anastasia P.; Faskhutdinova, Gulnaz; Kittler, Ellen L. W.; Moliaka, Yuri K. (2009). “Genotype Analysis Identifies the Cause of the ‘Royal Disease'”. Science. 326 (5954): 817. Bibcode:2009Sci…326..817R. doi:10.1126/science.1180660. PMID 19815722. S2CID 206522975.
- ^ “Hemophilia B”. Archived 2007-12-01 at the Wayback Machine National Hemophilia Foundation.
- ^ Carlo Gelmetti; Caputo, Ruggero (2002). Pediatric Dermatology and Dermatopathology: A Concise Atlas. T&F STM. p. 160. ISBN 1-84184-120-X.
- ^ “X-linked Agammaglobulinemia: Immunodeficiency Disorders: Merck Manual Professional”. Archived from the original on 2008-02-18. Retrieved 2008-03-01.
- ^ “Diseases Treated at St. Jude”. stjude.org. Archived from the original on 15 August 2007. Retrieved 3 May 2018.
- ^ “Favism – Doctor”. patient.info. Archived from the original on 21 November 2017. Retrieved 3 May 2018.
X-linked intellectual disability
X-linked intellectual disability refers to medical disorders associated with X-linked recessive inheritance that result in intellectual disability.
As with most X-linked disorders, males are more heavily affected than females.[1] Females with one affected X chromosome and one normal X chromosome tend to have milder symptoms.
Unlike many other types of intellectual disability, the genetics of these conditions are relatively well understood.[2][3] It has been estimated there are ~200 genes involved in this syndrome; of these ~100 have been identified.[4] Many of these genes are found on the short ‘p’ arm of the chromosome, and duplications at Xp11.2 are associated with the syndromic form of the condition.[5][6]
X-linked intellectual disability accounts for ~16% of all cases of intellectual disability in males.[7]
Syndromes
Several X-linked syndromes include intellectual disability as part of the presentation. These include:
- Coffin–Lowry syndrome
- DDX3X syndrome
- MASA syndrome
- MECP2 duplication syndrome
- Mental retardation and microcephaly with pontine and cerebellar hypoplasia
- X-linked alpha thalassemia mental retardation syndrome
List of genes
Following is a list of genes located on the X chromosome and linked to intellectual disability. There are also several loci that have not been associated with a specific gene.
- IQSEC2: encodes an exchange factor for the Arf family of small GTP binding proteins, involved in the formation of secretory vesicles.[8]
- TM4SF2: is a member of the 4 transmembrane domains family of proteins (tetraspanins, see TSPAN7). This gene is also associated with neuropsychiatric diseases such as Huntington’s chorea.[9]
- AP1S2: AP-1 complex subunit sigma-2.[10][11] Adaptor protein complex 1 is found on the cytoplasmic face of vesicles located at the Golgi complex, where it mediates both the recruitment of clathrin to the membrane and the recognition of sorting signals within the cytosolic tails of transmembrane receptors.
- ACSL4: Long-chain-fatty-acid—CoA ligase 4 is an enzyme of the long-chain fatty-acid–coenzyme A ligase family. It converts free long-chain fatty acids into fatty acyl–CoA esters, and thereby play a key role in lipid biosynthesis and fatty acid degradation.[12] This isozyme preferentially utilizes arachidonate as substrate.
- ZNF41: Zinc finger protein 41 is a likely zinc finger family transcription factor.[13]
- DLG3: Disks large homolog 3, also named neuroendocrine-DLG or synapse-associated protein 102 (SAP-102).[14] DLG3 is a member of the membrane-associated guanylate kinase (MAGUK) superfamily.
- FTSJ1: Transfert RNA methyltransferase 1 is a member of the S-adenosylmethionine-binding protein family. This nucleolar protein is involved in the processing and modification of tRNA.[15][16]
- GDI1: RabGDI alpha makes a complex with geranylgeranylated small GTP-binding proteins of the Rab family and keeps them in the cytosol.
- MECP2: methyl CpG binding protein 2 is a transcription regulator, which represses transcription from methylated gene promoters. It appears to be essential for the normal function of nerve cells.[17] In contrast to other MBD family members, MECP2 is X-linked and subject to X inactivation. MECP2 gene mutations are the cause of most cases of Rett syndrome, a progressive neurologic developmental disorder and one of the most common causes of intellectual disability in women.
- ARX: Aristaless related homeobox, is a protein associated with intellectual disability and lissencephaly. This gene is a homeobox-containing gene expressed during development. The expressed protein contains two conserved domains, a C-peptide (or aristaless domain) and the prd-like class homeobox domain. It is a member of the group-II aristaless-related protein family whose members are expressed primarily in the central and/or peripheral nervous system. This gene is involved in CNS and pancreas development. Mutations in this gene cause X-linked intellectual disability and epilepsy.[18]
- KDM5C: Lysine-specific demethylase 5C is an enzyme that in humans is encoded by the KDM5C gene a member of the SMCY homolog family and encodes a protein with one ARID domain, one JmjC domain, one JmjN domain and two PHD-type zinc fingers. The DNA-binding motifs suggest this protein is involved in the regulation of transcription and chromatin remodeling.[19]
- PHF8: PHD finger protein 8 belongs to the family of ferrous iron and 2-oxoglutarate dependent oxygenases,[20] and is a histone lysine demethylase with selectivity for the di-and monomethyl states.[21]
- FMR2: Fragile mental retardation 2 (FMR2: synonym AFF2),[22] the protein belongs to the AFF family which currently has four members: AFF1/AF4, AFF2/FMR2, AFF3/LAF4 and AFF4/AF5q31.[23] All AFF proteins are localized in the nucleus and have a role as transcriptional activators with a positive action on RNA elongation. AFF2/FMR2, AFF3/LAF4 and AFF4/AF5q31 localize in nuclear speckles (subnuclear structures considered to be storage/modification sites of pre-mRNA splicing factors) and are able to bind RNA with a high apparent affinity for the G-quadruplex structure. They appear to modulate alternative splicing via the interaction with the G-quadruplex RNA-forming structure.
- Slc6a8: Creatine transporter is a protein that is required for creatine to enter the cell. Creatine is essential for maintaining ATP levels in cells with a high energy demand.[24]
- GSPT2[25]
- MAGED1[26]
- UBE2A[27]
See also
- Xp11.2 Duplication
- Cell membrane protein disorders (other than Cell surface receptor, enzymes, and cytoskeleton)
- Deficiencies of intracellular signaling peptides and proteins
- Genetic disorders relating to deficiencies of transcription factor or coregulators
- Inherited disorders of trafficking / vesicular transport proteins
References
- ^ “Fragile X Syndrome – X-linked Mental Retardation and Macroorchidism”. International Birth Defect Information Systems. Retrieved 2010-12-10.
- ^ Ropers HH, Hamel BC (January 2005). “X-linked mental retardation”. Nature Reviews. Genetics. 6 (1): 46–57. doi:10.1038/nrg1501. PMID 15630421. S2CID 427210.
- ^ Lugtenberg D, Veltman JA, van Bokhoven H (September 2007). “High-resolution genomic microarrays for X-linked mental retardation”. Genetics in Medicine. 9 (9): 560–565. doi:10.1097/GIM.0b013e318149e647. PMID 17873643.
- ^ Stevenson RE, Schwartz CE (2009). “X-linked intellectual disability: unique vulnerability of the male genome”. Developmental Disabilities Research Reviews. 15 (4): 361–368. doi:10.1002/ddrr.81. PMID 20014364.
- ^ “OMIM Entry – # 300705 – CHROMOSOME Xp11.22 DUPLICATION SYNDROME”. omim.org. Retrieved 2018-03-09.
- ^ “Microduplication Xp11.22-p11.23 syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program”. rarediseases.info.nih.gov. Retrieved 2018-03-09.
- ^ Stevenson RE, Schwartz CE (2009). “X-linked intellectual disability: unique vulnerability of the male genome”. Developmental Disabilities Research Reviews. 15 (4): 361–368. doi:10.1002/ddrr.81. PMID 20014364.
- ^ Shoubridge C, Tarpey PS, Abidi F, Ramsden SL, Rujirabanjerd S, Murphy JA, et al. (June 2010). “Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability”. Nature Genetics. 42 (6): 486–488. doi:10.1038/ng.588. PMC 3632837. PMID 20473311.
- ^ Abidi FE, Holinski-Feder E, Rittinger O, Kooy F, Lubs HA, Stevenson RE, Schwartz CE (June 2002). “A novel 2 bp deletion in the TM4SF2 gene is associated with MRX58”. Journal of Medical Genetics. 39 (6): 430–433. doi:10.1136/jmg.39.6.430. PMC 1735161. PMID 12070254.
- ^ Tarpey PS, Stevens C, Teague J, Edkins S, O’Meara S, Avis T, et al. (December 2006). “Mutations in the gene encoding the Sigma 2 subunit of the adaptor protein 1 complex, AP1S2, cause X-linked mental retardation”. American Journal of Human Genetics. 79 (6): 1119–1124. doi:10.1086/510137. PMC 1698718. PMID 17186471.
- ^ “AP1S2 adaptor-related protein complex 1, sigma 2 subunit”. Entrez Gene. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Piccini M, Vitelli F, Bruttini M, Pober BR, Jonsson JJ, Villanova M, et al. (February 1998). “FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation”. Genomics. 47 (3): 350–358. doi:10.1006/geno.1997.5104. PMID 9480748.
- ^ Franzè A, Archidiacono N, Rocchi M, Marino M, Grimaldi G (April 1991). “Isolation and expression analysis of a human zinc finger gene (ZNF41) located on the short arm of the X chromosome”. Genomics. 9 (4): 728–736. doi:10.1016/0888-7543(91)90367-N. PMID 2037297.
- ^ Stathakis DG, Lee D, Bryant PJ (April 1998). “DLG3, the gene encoding human neuroendocrine Dlg (NE-Dlg), is located within the 1.8-Mb dystonia-parkinsonism region at Xq13.1”. Genomics. 49 (2): 310–313. doi:10.1006/geno.1998.5243. PMID 9598320.
- ^ Ramser J, Winnepenninckx B, Lenski C, Errijgers V, Platzer M, Schwartz CE, et al. (September 2004). “A splice site mutation in the methyltransferase gene FTSJ1 in Xp11.23 is associated with non-syndromic mental retardation in a large Belgian family (MRX9)”. Journal of Medical Genetics. 41 (9): 679–683. doi:10.1136/jmg.2004.019000. PMC 1735884. PMID 15342698.
- ^ Guy MP, Phizicky EM (January 2015). “Conservation of an intricate circuit for crucial modifications of the tRNAPhe anticodon loop in eukaryotes”. RNA. 21 (1): 61–74. doi:10.1261/rna.047639.114. PMC 4274638. PMID 25404562.
- ^ Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY (May 2008). “MeCP2, a key contributor to neurological disease, activates and represses transcription”. Science. 320 (5880): 1224–1229. Bibcode:2008Sci…320.1224C. doi:10.1126/science.1153252. PMC 2443785. PMID 18511691.
- ^ Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, et al. (April 2002). “ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation”. Human Molecular Genetics. 11 (8): 981–991. doi:10.1093/hmg/11.8.981. PMID 11971879.
- ^ Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, et al. (February 2005). “Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation”. American Journal of Human Genetics. 76 (2): 227–236. doi:10.1086/427563. PMC 1196368. PMID 15586325.
- ^ Loenarz C, Schofield CJ (March 2008). “Expanding chemical biology of 2-oxoglutarate oxygenases”. Nature Chemical Biology. 4 (3): 152–156. doi:10.1038/nchembio0308-152. PMID 18277970.
- ^ Loenarz C, Ge W, Coleman ML, Rose NR, Cooper CD, Klose RJ, et al. (January 2010). “PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nepsilon-dimethyl lysine demethylase”. Human Molecular Genetics. 19 (2): 217–222. doi:10.1093/hmg/ddp480. PMC 4673897. PMID 19843542.
- ^ Stettner GM, Shoukier M, Höger C, Brockmann K, Auber B (August 2011). “Familial intellectual disability and autistic behavior caused by a small FMR2 gene deletion”. American Journal of Medical Genetics. Part A. 155A (8): 2003–2007. doi:10.1002/ajmg.a.34122. PMID 21739600. S2CID 9568277.
- ^ Melko M, Douguet D, Bensaid M, Zongaro S, Verheggen C, Gecz J, Bardoni B (May 2011). “Functional characterization of the AFF (AF4/FMR2) family of RNA-binding proteins: insights into the molecular pathology of FRAXE intellectual disability”. Human Molecular Genetics. 20 (10): 1873–1885. doi:10.1093/hmg/ddr069. PMID 21330300.
- ^ Cecil KM, Salomons GS, Ball WS, Wong B, Chuck G, Verhoeven NM, et al. (March 2001). “Irreversible brain creatine deficiency with elevated serum and urine creatine: a creatine transporter defect?”. Annals of Neurology. 49 (3): 401–404. doi:10.1002/ana.79. PMID 11261517. S2CID 38756630.
- ^ Grau C, Starkovich M, Azamian MS, Xia F, Cheung SW, Evans P, et al. (2017). “Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability”. PLOS ONE. 12 (4): e0175962. Bibcode:2017PLoSO..1275962G. doi:10.1371/journal.pone.0175962. PMC 5393878. PMID 28414775.
- ^ Grau C, Starkovich M, Azamian MS, Xia F, Cheung SW, Evans P, et al. (2017). “Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability”. PLOS ONE. 12 (4): e0175962. Bibcode:2017PLoSO..1275962G. doi:10.1371/journal.pone.0175962. PMC 5393878. PMID 28414775.
- ^ Czeschik JC, Bauer P, Buiting K, Dufke C, Guillén-Navarro E, Johnson DS, et al. (September 2013). “X-linked intellectual disability type Nascimento is a clinically distinct, probably underdiagnosed entity”. Orphanet Journal of Rare Diseases. 8: 146. doi:10.1186/1750-1172-8-
Leave a Reply