Protoporphyrin ferrochelatase (EC 4.98.1.1, formerly EC 4.99.1.1, or ferrochelatase; systematic name protoheme ferro-lyase (protoporphyrin-forming)) is an enzyme encoded by the FECH gene in humans.
Ferrochelatase catalyses the eighth and terminal step in the biosynthesis of heme, converting protoporphyrin IX into heme B.
It catalyses the reaction:
protoheme + 2 H+ = protoporphyrin + Fe2+

Function
Ferrochelatase catalyzes the insertion of ferrous iron into protoporphyrin IX in the heme biosynthesis pathway to form heme B. The enzyme is localized to the matrix-facing side of the inner mitochondrial membrane. Ferrochelatase is the best known member of a family of enzymes that add divalent metal cations to tetrapyrrole structures.
- Lecerof, D.; Fodje, M.; Hansson, A.; Hansson, M.; Al-Karadaghi, S. (March 2000). “Structural and mechanistic basis of porphyrin metallation by ferrochelatase”. Journal of Molecular Biology. 297 (1): 221–232. doi:10.1006/jmbi.2000.3569. PMID 10704318.
For example, magnesium chelatase adds magnesium to protoporphyrin IX in the first step of bacteriochlorophyll biosynthesis.
- Leeper, F. J. (1985). “The biosynthesis of porphyrins, chlorophylls, and vitamin B12”. Natural Product Reports. 2 (1): 19–47. doi:10.1039/NP9850200019. PMID 3895052.
Bacteriochlorophyll Notes
Bacteriochlorophylls (BChl) are photosynthetic pigments that occur in various phototrophicbacteria. They were discovered by C. B. van Niel in 1932.[Niel, C. B. (1932). “On the morphology and physiology of the purple and green sulphur bacteria”. Archiv für Mikrobiologie. 3: 1–112. doi:10.1007/BF00454965. S2CID 19597530.] They are related to chlorophylls, which are the primary pigments in plants, algae, and cyanobacteria. Organisms that contain bacteriochlorophyll conduct photosynthesis to sustain their energy requirements, but the process is anoxygenic and does not produce oxygen as a byproduct. They use wavelengths of light not absorbed by plants or cyanobacteria. Replacement of Mg2+ with protons gives bacteriophaeophytin (BPh), the phaeophytin form.
There are a large number of known bacteriochlorophylls[Senge, Mathias O.; Smith, Kevin M. (2004). “Biosynthesis and Structures of the Bacteriochlorophylls”. Anoxygenic Photosynthetic Bacteria. Advances in Photosynthesis and Respiration. Vol. 2. pp. 137–151. doi:10.1007/0-306-47954-0_8. ISBN 0-7923-3681-X.][Chew, Aline Gomez Maqueo; Bryant, Donald A. (2007). “Chlorophyll Biosynthesis in Bacteria: The Origins of Structural and Functional Diversity”. Annual Review of Microbiology. 61: 113–129. doi:10.1146/annurev.micro.61.080706.093242. PMID 17506685.] but all have features in common since the biosynthetic pathway involves chlorophyllide a (Chlide a) as an intermediate.[Willows, Robert D. (2003). “Biosynthesis of chlorophylls from protoporphyrin IX”. Natural Product Reports. 20 (6): 327–341. doi:10.1039/B110549N. PMID 12828371.]
Chlorin-cored BChls (c to f) are produced by a series of enzymatic modifications on the sidechain of Chlide a, much like how Chl b, d, e are made. The bacteriochlorin-cored BChls a, b, g require a unique step to reduce the double bound between C7 and C8, which is performed by Chlorophyllide a reductase (COR).[Chew, Aline Gomez Maqueo; Bryant, Donald A. (2007). “Chlorophyll Biosynthesis in Bacteria: The Origins of Structural and Functional Diversity”. Annual Review of Microbiology. 61: 113–129. doi:10.1146/annurev.micro.61.080706.093242. PMID 17506685.]
Isobacteriochlorins, in contrast, are biosynthesised from uroporphyrinogen III in a separate pathway that leads, for example, to siroheme, cofactor F430 and cobalamin. The common intermediate is sirohydrochlorin.[Battersby, Alan R. (2000). “Tetrapyrroles: The pigments of life: A Millennium review”. Natural Product Reports. 17 (6): 507–526. doi:10.1039/B002635M. PMID 11152419.]
Heme B is an essential cofactor in many proteins and enzymes. In particular, heme b plays a key role as the oxygen carrier in hemoglobin in red blood cells and myoglobin in muscle cells. Furthermore, heme B is found in cytochrome b, a key component in Q-cytochrome c oxidoreductase (complex III) in oxidative phosphorylation.
- Berg, Jeremy; Tymoczko, John; Stryer, Lubert (2012). Biochemistry (7th ed.). New York: W.H. Freeman. ISBN 9781429229364.
Cytochrome b Notes
Cytochrome b within both molecular and cell biology, is a protein found in the mitochondria of eukaryotic cells. It functions as part of the electron transport chain and is the main subunit of transmembrane cytochrome bc1 and b6f complexes.[Howell N (August 1989). “Evolutionary conservation of protein regions in the proton motive cytochrome b and their possible roles in redox catalysis”. J. Mol. Evol. 29 (2): 157–69. Bibcode:1989JMolE..29..157H. doi:10.1007/BF02100114. PMID 2509716. S2CID 7298013.][Esposti MD, De Vries S, Crimi M, Ghelli A, Patarnello T, Meyer A (July 1993). “Mitochondrial cytochrome b: evolution and structure of the protein” (PDF). Biochim. Biophys. Acta. 1143 (3): 243–71. doi:10.1016/0005-2728(93)90197-N. PMID 8329437.]
Cytochrome b Function
In the mitochondrion of eukaryotes and in aerobic prokaryotes, cytochrome b is a component of respiratory chain complex III (EC 1.10.2.2) — also known as the bc1 complex or ubiquinol-cytochrome c reductase. In plant chloroplasts and cyanobacteria, there is an analogous protein, cytochrome b6, a component of the plastoquinone-plastocyanin reductase (EC 1.10.99.1), also known as the b6f complex. These complexes are involved in electron transport, the pumping of protons to create a proton-motive force (PMF). This proton gradient is used for the generation of ATP. These complexes play a vital role in cells.[Blankenship, Robert (2009). Molecular Mechanisms of Photosynthesis. Blackwell Publishing. pp. 124–132.]
Cytochrome b Structure
Cytochrome b/b6[Howell N (1989). “Evolutionary conservation of protein regions in the protonmotive cytochrome b and their possible roles in redox catalysis”. J. Mol. Evol. 29 (2): 157–169. Bibcode:1989JMolE..29..157H. doi:10.1007/BF02100114. PMID 2509716. S2CID 7298013.][Esposti MD, Crimi M, Ghelli A, Patarnello T, Meyer A, De Vries S (1993). “Mitochondrial cytochrome b: evolution and structure of the protein” (PDF). Biochim. Biophys. Acta. 1143 (3): 243–271. doi:10.1016/0005-2728(93)90197-N. PMID 8329437.] is an integral membrane protein of approximately 400 amino acid residues that probably has 8 transmembrane segments. In plants and cyanobacteria, cytochrome b6 consists of two protein subunits encoded by the petB and petD genes. Cytochrome b/b6 non-covalently binds two heme groups, known as b562 and b566. Four conserved histidine residues are postulated to be the ligands of the iron atoms of these two heme groups.
Cytochrome b Use in phylogenetics
Cytochrome b is commonly used as a region of mitochondrial DNA for determining phylogenetic relationships between organisms, due to its sequence variability. It is considered to be most useful in determining relationships within families and genera. Comparative studies involving cytochrome b have resulted in new classification schemes and have been used to assign newly described species to a genus as well as to deepen the understanding of evolutionary relationships.[Castresana, J. (2001). “Cytochrome b Phylogeny and the Taxonomy of Great Apes and Mammals”. Molecular Biology and Evolution. 18 (4): 465–471. doi:10.1093/oxfordjournals.molbev.a003825. PMID 11264397.]
Cytochrome b Clinical significance
Mutations in cytochrome b primarily result in exercise intolerance in human patients; though more rare, severe multi-system pathologies have also been reported.[Blakely EL, Mitchell AL, Fisher N, Meunier B, Nijtmans LG, Schaefer AM, Jackson MJ, Turnbull DM, Taylor RW (July 2005). “A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast”. FEBS J. 272 (14): 3583–92. doi:10.1111/j.1742-4658.2005.04779.x. PMID 16008558. S2CID 13938075.]
Single-point mutations in cytochrome b of Plasmodium falciparum and P. berghei are associated with resistance to the anti-malarial drug atovaquone.[Siregar JE, Syafruddin D, Matsuoka H, Kita K, Marzuki S (June 2008). “Mutation underlying resistance of Plasmodium berghei to atovaquone in the quinone binding domain 2 (Qo(2)) of the cytochrome b gene”. Parasitology International. 57 (2): 229–32. doi:10.1016/j.parint.2007.12.002. PMID 18248769]
Cytochrome b Human genes
Human genes encoding cytochrome b proteins include:
- CYB5A – cytochrome b5 type A (microsomal)
- Cytochrome b5, form A (gene name CYB5A), is a human microsomal cytochrome b5.[“Entrez Gene: CYB5A Cytochrome b5, form A”.] Cytochrome b5 is a membrane bound hemoprotein which functions as an electron carrier for several membrane bound oxygenases. It has two isoforms produced by alternative splicing. Isoform 1 is bound to the cytoplasmic side of the endoplasmic reticulum. It has a C-terminal transmembrane alpha-helix. Isoform 2 was found in cytoplasm. Defects in CYB5A are the cause of type IV hereditary methemoglobinemia.
- CYB5B – cytochrome b5 type B (outer mitochondrial membrane)
- Redirects to MT-CYB
- The MT-CYB gene is located on the p arm of mitochondrial DNA in position 12 and spans 1,140 base pairs.[ “Entrez Gene: CYTB cytochrome b”.] The gene produces a 42.7 kDa protein named cytochrome b composed of 380 amino acids.[Zong NC, Li H, Li H, Lam MP, Jimenez RC, Kim CS, Deng N, Kim AK, Choi JH, Zelaya I, Liem D, Meyer D, Odeberg J, Fang C, Lu HJ, Xu T, Weiss J, Duan H, Uhlen M, Yates JR, Apweiler R, Ge J, Hermjakob H, Ping P (Oct 2013). “Integration of cardiac proteome biology and medicine by a specialized knowledgebase”. Circulation Research. 113 (9): 1043–53. doi:10.1161/CIRCRESAHA.113.301151. PMC 4076475. PMID 23965338.][“cytochrome b”. Cardiac Organellar Protein Atlas Knowledgebase (COPaKB).]Cytochrome b is an integral membrane protein with hydrophobic properties. The catalytic core of the enzyme is composed of eight transmembrane helices, the iron-sulfur protein, and cytochrome c1.[Fragaki K, Procaccio V, Bannwarth S, Serre V, O’Hearn S, Potluri P, Augé G, Casagrande F, Caruba C, Lambert JC, Paquis-Flucklinger V (September 2009). “A neonatal polyvisceral failure linked to a de novo homoplasmic mutation in the mitochondrially encoded cytochrome b gene”. Mitochondrion. 9 (5): 346–52. doi:10.1016/j.mito.2009.06.002. PMID 19563916.]Cytochrome b is a fundamental component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c.[“UniProtKB – P00156 (CYB_HUMAN)”. The UniProt Consortium.] The structure of the complex is a symmetric homodimer. It is composed of eleven structural subunits, including one mitochondrial genome encoded cytochrome b and ten other nucleus encoded subunits. These subunits include three respiratory subunits (MT-CYB, CYC1 and UQCRFS1), two core proteins (UQCRC1 and UQCRC2) and six low-molecular weight proteins (UQCRH/QCR6, UQCRB/QCR7, UQCRQ/QCR8, UQCR10/QCR9, UQCR11/QCR10 and a cleavage product of UQCRFS1). The total molecular mass of the complex is about 450 kDa.[Gil Borlado MC, Moreno Lastres D, Gonzalez Hoyuela M, Moran M, Blazquez A, Pello R, Marin Buera L, Gabaldon T, Garcia Peñas JJ, Martín MA, Arenas J, Ugalde C (September 2010). “Impact of the mitochondrial genetic background in complex III deficiency”. PLOS ONE. 5 (9): e12801. Bibcode:2010PLoSO…512801G. CiteSeerX 10.1.1.350.7243. doi:10.1371/journal.pone.0012801. PMC 2941448. PMID 20862300.][“UniProtKB – P00156 (CYB_HUMAN)”. The UniProt Consortium.]
- CYBASC3 – cytochrome b, ascorbate dependent 3
- MT-CYB – mitochondrially encoded cytochrome b
- Cytochrome b is a protein that in humans is encoded by the MT-CYB gene.[ “Entrez Gene: CYTB cytochrome b”.] Its gene product is a subunit of the respiratory chain protein ubiquinol–cytochrome c reductase (UQCR, complex III or cytochrome bc1 complex), which consists of the products of one mitochondrially encoded gene, MT-CYB (mitochondrial cytochrome b), and ten nuclear genes—UQCRC1, UQCRC2, CYC1, UQCRFS1 (Rieske protein), UQCRB, “11kDa protein”, UQCRH (cyt c1 Hinge protein), Rieske protein presequence, “cyt c1 associated protein”, and Rieske-associated protein.
- The mitochondrial cytochrome b is fundamental for the assembly and function of Complex III of the mitochondrial respiratory chain.[Massie R, Wong LJ, Milone M (July 2010). “Exercise intolerance due to cytochrome b mutation”. Muscle & Nerve. 42 (1): 136–40. doi:10.1002/mus.21649. PMID 20544923. S2CID 23759055.] Complex III is responsible for the catalysis of electron transfer from coenzyme Q to cytochrome c in the mitochondrial respiratory chain by translocating protons concomitantly across the inner membrane of the mitochondria.[12][“UniProtKB – P00156 (CYB_HUMAN)”. The UniProt Consortium.] The transfer of electrons then contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis.[“UniProtKB – P00156 (CYB_HUMAN)”. The UniProt Consortium.]
- Mutations in MT-CYB can result in mitochondrial deficiencies and associated disorders. It is majorly associated with a complex III deficiency, a deficiency in an enzyme complex which catalyzes electron transfer from coenzyme Q to cytochrome c in the mitochondrial respiratory chain. A complex III deficiency can result in a highly variable phenotype depending on which tissues are affected.[“UniProtKB – P00156 (CYB_HUMAN)”. The UniProt Consortium.] Most frequent clinical manifestations include progressive exercise intolerance and cardiomyopathy. Occasional multisystem disorders accompanied by exercise intolerance may arise as well, in forms of deafness, mental retardation, retinitis pigmentosa, cataract, growth retardation, and epilepsy.[“UniProtKB – P00156 (CYB_HUMAN)”. The UniProt Consortium.] Other phenotypes include mitochondrial encephalomyopathy, mitochondrial myopathy, Leber hereditary optic neuropathy, muscle weakness, myoglobinuria, blood acidosis, renal tubulopathy, and more.[“UniProtKB – P00156 (CYB_HUMAN)”. The UniProt Consortium.][Gil Borlado MC, Moreno Lastres D, Gonzalez Hoyuela M, Moran M, Blazquez A, Pello R, Marin Buera L, Gabaldon T, Garcia Peñas JJ, Martín MA, Arenas J, Ugalde C (September 2010). “Impact of the mitochondrial genetic background in complex III deficiency” PLOS ONE. 5 (9):e12801. Bibcode:2010PLoSO…512801G. CiteSeerX 10.1.1.350.7243. doi:10.1371/journal.pone.0012801. PMC 2941448. PMID 20862300] Complex III deficiency is known to be rare among mitochondrial diseases and may follow a maternal or mendelian mode of inheritance due to its duality of genetic origin.[Fragaki K, Procaccio V, Bannwarth S, Serre V, O’Hearn S, Potluri P, Augé G, Casagrande F, Caruba C, Lambert JC, Paquis-Flucklinger V (September 2009). “A neonatal polyvisceral failure linked to a de novo homoplasmic mutation in the mitochondrially encoded cytochrome b gene”. Mitochondrion. 9 (5): 346–52. doi:10.1016/j.mito.2009.06.002. PMID 19563916.]
Cytochrome b Fungicide target
Cyt b is targeted by the QoI class of fungicides, Fungicide Resistance Action Committee group 11. The cyt b mutations G143A and F129L provide resistance against the main body of group 11, although G143A does not work against metyltetraprole (11A).[FRAC (Fungicide Resistance Action Committee) (March 2021). “FRAC Code List ©*2021: Fungal control agents sorted by cross resistance pattern and mode of action (including coding for FRAC Groups on product labels)” (PDF).] G143A is significant in Botrytis cinerea in California strawberry production.[Petrasch, Stefan; Knapp, Steven J.; van Kan, Jan A. L.; Blanco‐Ulate, Barbara (2019-04-04). “Grey mould of strawberry, a devastating disease caused by the ubiquitous necrotrophic fungal pathogen Botrytis cinerea“. Molecular Plant Pathology. British Society for Plant Pathology (W-B). 20 (6): 877–892. doi:10.1111/mpp.12794. ISSN 1464-6722. PMC 6637890. PMID 30945788. S2CID 93002697. Cosseboom, Scott D.; Ivors, Kelly L.; Schnabel, Guido; Bryson, Patricia K.; Holmes, Gerald J. (2019). “Within-Season Shift in Fungicide Resistance Profiles of Botrytis cinerea in California Strawberry Fields”. Plant Disease. American Phytopathological Society. 103 (1): 59–64. doi:10.1094/pdis-03-18-0406-re. ISSN 0191-2917. PMID 30422743. S2CID 205345358.]
- Qo inhibitors (QoI),[ “quinone outside inhibitor (CHEBI:141153)”. The European Bioinformatics Institute < EMBL-EBI. Retrieved 2021-02-20.] or quinone outside inhibitors, are a group of fungicides used in agriculture. Some of these fungicides are among the most popular in the world.[ Hawkins, N.J.; Fraaije, B.A. (2018-08-25). “Fitness Penalties in the Evolution of Fungicide Resistance”. Annual Review of Phytopathology. Annual Reviews. 56 (1): 339–360. doi:10.1146/annurev-phyto-080417-050012. ISSN 0066-4286.] QoI are chemical compounds which act at the quinol outer binding site of the cytochrome bc1 complex.
- Most QoI common names end in -strobin and so are often called strobs.[FRAC (Fungicide Resistance Action Committee) (March 2021). “FRAC Code List ©*2021: Fungal control agents sorted by cross resistance pattern and mode of action (including coding for FRAC Groups on product labels)” (PDF). pp. 1–17.] QoI’s are the resulting fusion of three fungicides families, the well-known family of strobilurins and two new families, represented by fenamidone and famoxadone. Some strobilurins are azoxystrobin, kresoxim-methyl, picoxystrobin, pyraclostrobin, and trifloxystrobin.
- QoI fungicides are used on a wide range of crops, such as cereals, vines, pome fruits, cucurbits, tomatoes, and potatoes.[citation needed]
- For example, they are used as fungicides for cereals, against Erysiphe graminis f.sp tritici responsible for the powdery mildew in wheat or against Septoria tritici, responsible for septoria leaf spot in wheat.[citation needed]
- They are also commonly used for vine culture, against Plasmopara viticola, responsible for downy mildew or in oïdium treatment.[citation needed]
- List
- QoIs:[FRAC (Fungicide Resistance Action Committee) (March 2021). “FRAC Code List ©*2021: Fungal control agents sorted by cross resistance pattern and mode of action (including coding for FRAC Groups on product labels)” (PDF). pp. 1–17.]
- QoI subgroup A:
Almost all these fungicides are in the same cross-resistance group (FRAC 11) and must be managed carefully to avoid the appearance of fungicide resistance. All group 11s are cross-resistant with each other.[FRAC (Fungicide Resistance Action Committee) (March 2021). “FRAC Code List ©*2021: Fungal control agents sorted by cross resistance pattern and mode of action (including coding for FRAC Groups on product labels)” (PDF). pp. 1–17.] Some fungicide resistance has been observed in many crop pathogens[ Zeng, F; Arnao, E; Zhang, G; Olaya, G; Wullschleger, J; Sierotzki, H; Ming, R; Bluhm, B. H; Bond, J. P; Fakhoury, A. M; Bradley, C. A (2015). “Characterization of Quinone Outside Inhibitor Fungicide Resistance in Cercospora sojina and Development of Diagnostic Tools for its Identification”. Plant Disease. 99 (4): 544–550. doi:10.1094/PDIS-05-14-0460-RE.] (such as in the case of wheat powdery mildew), so the application of QoI products should respect effective rates and intervals to provides time and space when the pathogen population is not influenced by the product selection pressure.[clarification needed][“Recommendations for QoI”. FRAC (Fungicide Resistance Action Committee). 2020-01-31. Retrieved 2021-06-16.] Resistance to group 11 is conferred by cytochrome b mutations G143A and F129L, and by other mechanisms.[FRAC (Fungicide Resistance Action Committee) (March 2021). “FRAC Code List ©*2021: Fungal control agents sorted by cross resistance pattern and mode of action (including coding for FRAC Groups on product labels)” (PDF). pp. 1–17.]
The tetrazolinones consist of only one molecule, metyltetraprole. This constitutes FRAC 11A. 11A is not cross-resistant with 11 resistance conferred by G143A.[3]
See also:
Cytochrome b References
- Howell N (August 1989). “Evolutionary conservation of protein regions in the proton motive cytochrome b and their possible roles in redox catalysis”. J. Mol. Evol. 29 (2): 157–69. Bibcode:1989JMolE..29..157H. doi:10.1007/BF02100114. PMID 2509716. S2CID 7298013.
- Esposti MD, De Vries S, Crimi M, Ghelli A, Patarnello T, Meyer A (July 1993). “Mitochondrial cytochrome b: evolution and structure of the protein” (PDF). Biochim. Biophys. Acta. 1143 (3): 243–71. doi:10.1016/0005-2728(93)90197-N. PMID 8329437.
- Blankenship, Robert (2009). Molecular Mechanisms of Photosynthesis. Blackwell Publishing. pp. 124–132.
- Howell N (1989). “Evolutionary conservation of protein regions in the protonmotive cytochrome b and their possible roles in redox catalysis”. J. Mol. Evol. 29 (2): 157–169. Bibcode:1989JMolE..29..157H. doi:10.1007/BF02100114. PMID 2509716. S2CID 7298013.
- Esposti MD, Crimi M, Ghelli A, Patarnello T, Meyer A, De Vries S (1993). “Mitochondrial cytochrome b: evolution and structure of the protein” (PDF). Biochim. Biophys. Acta. 1143 (3): 243–271. doi:10.1016/0005-2728(93)90197-N. PMID 8329437.
- Castresana, J. (2001). “Cytochrome b Phylogeny and the Taxonomy of Great Apes and Mammals”. Molecular Biology and Evolution. 18 (4): 465–471. doi:10.1093/oxfordjournals.molbev.a003825. PMID 11264397.
- Blakely EL, Mitchell AL, Fisher N, Meunier B, Nijtmans LG, Schaefer AM, Jackson MJ, Turnbull DM, Taylor RW (July 2005). “A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast”. FEBS J. 272 (14): 3583–92. doi:10.1111/j.1742-4658.2005.04779.x. PMID 16008558. S2CID 13938075.
- Siregar JE, Syafruddin D, Matsuoka H, Kita K, Marzuki S (June 2008). “Mutation underlying resistance of Plasmodium berghei to atovaquone in the quinone binding domain 2 (Qo(2)) of the cytochrome b gene”. Parasitology International. 57 (2): 229–32. doi:10.1016/j.parint.2007.12.002. PMID 18248769.
- FRAC (Fungicide Resistance Action Committee) (March 2021). “FRAC Code List ©*2021: Fungal control agents sorted by cross resistance pattern and mode of action (including coding for FRAC Groups on product labels)” (PDF).
- Petrasch, Stefan; Knapp, Steven J.; van Kan, Jan A. L.; Blanco‐Ulate, Barbara (2019-04-04). “Grey mould of strawberry, a devastating disease caused by the ubiquitous necrotrophic fungal pathogen Botrytis cinerea”. Molecular Plant Pathology. British Society for Plant Pathology (W-B). 20 (6): 877–892. doi:10.1111/mpp.12794. ISSN 1464-6722. PMC 6637890. PMID 30945788. S2CID 93002697.
- Cosseboom, Scott D.; Ivors, Kelly L.; Schnabel, Guido; Bryson, Patricia K.; Holmes, Gerald J. (2019). “Within-Season Shift in Fungicide Resistance Profiles of Botrytis cinerea in California Strawberry Fields”. Plant Disease. American Phytopathological Society. 103 (1): 59–64. doi:10.1094/pdis-03-18-0406-re. ISSN 0191-2917. PMID 30422743. S2CID 205345358.
Cytochrome b External links
- Cytochromes+b at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
| Proteins that contain heme (hemoproteins) |
|---|
Cytochrome b Categories:
Coenzyme Q : cytochrome c – oxidoreductase Notes
The coenzyme Q : cytochrome c – oxidoreductase, sometimes called the cytochrome bc1 complex, and at other times complex III, is the third complex in the electron transport chain (EC 1.10.2.2), playing a critical role in biochemical generation of ATP (oxidative phosphorylation). Complex III is a multisubunit transmembrane protein encoded by both the mitochondrial (cytochrome b) and the nuclear genomes (all other subunits). Complex III is present in the mitochondria of all animals and all aerobic eukaryotes and the inner membranes of most eubacteria. Mutations in Complex III cause exercise intolerance as well as multisystem disorders. The bc1 complex contains 11 subunits, 3 respiratory subunits (cytochrome B, cytochrome C1, Rieske protein), 2 core proteins and 6 low-molecular weight proteins.
Ubiquinol—cytochrome-c reductase catalyzes the chemical reaction
QH2 + 2 ferricytochrome c ⇌
Thus, the two substrates of this enzyme are quinol (QH2) and ferri- (Fe3+) cytochrome c, whereas its 3 products are quinone (Q), ferro- (Fe2+) cytochrome c, and H+.
This enzyme belongs to the family of oxidoreductases, specifically those acting on diphenols and related substances as donor with a cytochrome as acceptor. This enzyme participates in oxidative phosphorylation. It has four cofactors: cytochrome c1, cytochrome b-562, cytochrome b-566, and a 2-Iron ferredoxin of the Rieske type.
Coenzyme Q : cytochrome c – oxidoreductase Nomenclature
The systematic name of this enzyme class is ubiquinol:ferricytochrome-c oxidoreductase. Other names in common use include:
- coenzyme Q-cytochrome c reductase,
- dihydrocoenzyme Q-cytochrome c reductase,
- reduced ubiquinone-cytochrome c reductase, complex III,
- (mitochondrial electron transport),
- ubiquinone-cytochrome c reductase,
- ubiquinol-cytochrome c oxidoreductase,
- reduced coenzyme Q-cytochrome c reductase,
- ubiquinone-cytochrome c oxidoreductase,
- reduced ubiquinone-cytochrome c oxidoreductase,
- mitochondrial electron transport complex III,
- ubiquinol-cytochrome c-2 oxidoreductase,
- ubiquinone-cytochrome b-c1 oxidoreductase,
- ubiquinol-cytochrome c2 reductase,
- ubiquinol-cytochrome c1 oxidoreductase,
- CoQH2-cytochrome c oxidoreductase,
- ubihydroquinol:cytochrome c oxidoreductase,
- coenzyme QH2-cytochrome c reductase, and
- QH2:cytochrome c oxidoreductase.
Coenzyme Q : cytochrome c – oxidoreductase Structure

Compared to the other major proton-pumping subunits of the electron transport chain, the number of subunits found can be small, as small as three polypeptide chains. This number does increase, and eleven subunits are found in higher animals.[ Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK (July 1998). “Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex”. Science. 281 (5373): 64–71. Bibcode:1998Sci…281…64I. doi:10.1126/science.281.5373.64. PMID 9651245.] Three subunits have prosthetic groups. The cytochrome b subunit has two b-type hemes (bL and bH), the cytochrome c subunit has one c-type heme (c1), and the Rieske Iron Sulfur Protein subunit (ISP) has a two iron, two sulfur iron-sulfur cluster (2Fe•2S).
Structures of complex III: PDB: 1KYO, PDB: 1L0L
Coenzyme Q – cytochrome c reductase – Composition of complex
In vertebrates the bc1 complex, or Complex III, contains 11 subunits: 3 respiratory subunits, 2 core proteins and 6 low-molecular weight proteins.[Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, et al. (1998). “Electron transfer by domain movement in cytochrome bc1”. Nature. 392 (6677): 677–84. Bibcode:1998Natur.392..677Z. doi:10.1038/33612. PMID 9565029. S2CID 4380033.][Hao GF, Wang F, Li H, Zhu XL, Yang WC, Huang LS, et al. (2012). “Computational discovery of picomolar Q(o) site inhibitors of cytochrome bc1 complex”. J Am Chem Soc. 134 (27): 11168–76. doi:10.1021/ja3001908. PMID 22690928.] Proteobacterial complexes may contain as few as three subunits.[Yang XH, Trumpower BL (1986). “Purification of a three-subunit ubiquinol-cytochrome c oxidoreductase complex from Paracoccus denitrificans”. J Biol Chem. 261 (26): 12282–9. doi:10.1016/S0021-9258(18)67236-9. PMID 3017970.]
Table of subunit composition of complex III
| No. | Subunit name | Human protein | Protein description from UniProt | Pfam family with Human protein |
|---|---|---|---|---|
| Respiratory subunit proteins | ||||
| 1 | MT-CYB / Cyt b | CYB_HUMAN | Cytochrome b | Pfam PF13631 |
| 2 | CYC1 / Cyt c1 | CY1_HUMAN | Cytochrome c1, heme protein, mitochondrial | Pfam PF02167 |
| 3 | Rieske / UCR1 | UCRI_HUMAN | Cytochrome b-c1 complex subunit Rieske, mitochondrial EC 1.10.2.2 | Pfam PF02921 , Pfam PF00355 |
| Core protein subunits | ||||
| 4 | QCR1 / SU1 | QCR1_HUMAN | Cytochrome b-c1 complex subunit 1, mitochondrial | Pfam PF00675, Pfam PF05193 |
| 5 | QCR2 / SU2 | QCR2_HUMAN | Cytochrome b-c1 complex subunit 2, mitochondrial | Pfam PF00675, Pfam PF05193 |
| Low-molecular weight protein subunits | ||||
| 6 | QCR6 / SU6 | QCR6_HUMAN | Cytochrome b-c1 complex subunit 6, mitochondrial | Pfam PF02320 |
| 7 | QCR7 / SU7 | QCR7_HUMAN | Cytochrome b-c1 complex subunit 7 | Pfam PF02271 |
| 8 | QCR8 / SU8 | QCR8_HUMAN | Cytochrome b-c1 complex subunit 8 | Pfam PF02939 |
| 9 | QCR9 / SU9 / UCRC | QCR9_HUMANa | Cytochrome b-c1 complex subunit 9 | Pfam PF09165 |
| 10 | QCR10 / SU10 | QCR10_HUMAN | Cytochrome b-c1 complex subunit 10 | Pfam PF05365 |
| 11 | QCR11 / SU11 | QCR11_HUMAN | Cytochrome b-c1 complex subunit 11 | Pfam PF08997 |
Coenzyme Q – cytochrome c reductase Reaction
It catalyzes the reduction of cytochrome c by oxidation of coenzyme Q (CoQ) and the concomitant pumping of 4 protons from the mitochondrial matrix to the intermembrane space:
QH2 + 2 cytochrome c (FeIII) + 2 H+
in → Q + 2 cytochrome c (FeII) + 4 H+
out
In the process called Q cycle,[Kramer DM, Roberts AG, Muller F, Cape J, Bowman MK (2004). “Q-cycle bypass reactions at the Qo site of the cytochrome bc1 (and related) complexes”. Quinones and Quinone Enzymes, Part B. Meth. Enzymol. Methods in Enzymology. Vol. 382. pp. 21–45. doi:10.1016/S0076-6879(04)82002-0. ISBN 978-0-12-182786-1. PMID 15047094.][Crofts AR (2004). “The cytochrome bc1 complex: function in the context of structure”. Annu. Rev. Physiol. 66: 689–733. doi:10.1146/annurev.physiol.66.032102.150251. PMID 14977419.] two protons are consumed from the matrix (M), four protons are released into the inter membrane space (IM) and two electrons are passed to cytochrome c.
Coenzyme Q – cytochrome c reductase Reaction mechanism

The reaction mechanism for complex III (cytochrome bc1, coenzyme Q: cytochrome C oxidoreductase) is known as the ubiquinone (“Q”) cycle. In this cycle four protons get released into the positive “P” side (inter membrane space), but only two protons get taken up from the negative “N” side (matrix). As a result, a proton gradient is formed across the membrane. In the overall reaction, two ubiquinols are oxidized to ubiquinones and one ubiquinone is reduced to ubiquinol. In the complete mechanism, two electrons are transferred from ubiquinol to ubiquinone, via two cytochrome c intermediates.
Overall:
- 2 x QH2 oxidised to Q
- 1 x Q reduced to QH2
- 2 x Cyt c reduced
- 4 x H+ released into intermembrane space
- 2 x H+ picked up from matrix
The reaction proceeds according to the following steps:
Round 1:
- Cytochrome b binds a ubiquinol and a ubiquinone.
- The 2Fe/2S center and BL heme each pull an electron off the bound ubiquinol, releasing two protons into the intermembrane space.
- One electron is transferred to cytochrome c1 from the 2Fe/2S centre, whilst another is transferred from the BL heme to the BH Heme.
- Cytochrome c1 transfers its electron to cytochrome c (not to be confused with cytochrome c1), and the BH Heme transfers its electron to a nearby ubiquinone, resulting in the formation of a ubisemiquinone.
- Cytochrome c diffuses. The first ubiquinol (now oxidised to ubiquinone) is released, whilst the semiquinone remains bound.
Round 2:
- A second ubiquinol is bound by cytochrome b.
- The 2Fe/2S center and BL heme each pull an electron off the bound ubiquinol, releasing two protons into the intermembrane space.
- One electron is transferred to cytochrome c1 from the 2Fe/2S centre, whilst another is transferred from the BL heme to the BH Heme.
- Cytochrome c1 then transfers its electron to cytochrome c, whilst the nearby semiquinone produced from round 1 picks up a second electron from the BH heme, along with two protons from the matrix.
- The second ubiquinol (now oxidised to ubiquinone), along with the newly formed ubiquinol are released.[ Ferguson SJ, Nicholls D, Ferguson S (2002). Bioenergetics (3rd ed.). San Diego: Academic. pp. 114–117. ISBN 978-0-12-518121-1.]
Coenzyme Q – cytochrome c reductase – Inhibitors of complex III
There are three distinct groups of Complex III inhibitors.
- Antimycin A binds to the Qi site and inhibits the transfer of electrons in Complex III from heme bH to oxidized Q (Qi site inhibitor).
- Myxothiazol and stigmatellin binds to the Qo site and inhibits the transfer of electrons from reduced QH2 to the Rieske Iron sulfur protein. Myxothiazol and stigmatellin bind to distinct but overlapping pockets within the Qo site.
- Myxothiazol binds nearer to cytochrome bL (hence termed a “proximal” inhibitor).
- Stigmatellin binds farther from heme bL and nearer the Rieske Iron sulfur protein, with which it strongly interacts.
Some have been commercialized as fungicides (the strobilurin derivatives, best known of which is azoxystrobin; QoI inhibitors) and as anti-malaria agents (atovaquone).
Also propylhexedrine inhibits cytochrome c reductase.[Holmes, J. H.; Sapeika, N; Zwarenstein, H (1975). “Inhibitory effect of anti-obesity drugs on NADH dehydrogenase of mouse heart homogenates”. Research Communications in Chemical Pathology and Pharmacology. 11 (4): 645–6. PMID 241101.]
- Propylhexedrine, commonly sold under the brand name Benzedrex, is an adrenergic alkylamine primarily utilized as a topical nasal decongestant.[ “COLD, COUGH, ALLERGY, BRONCHODILATOR, AND ANTIASTHMATIC DRUG PRODUCTS FOR OVER-THE-COUNTER HUMAN USE”. FDA. 7 June 2023. Archived from the original on 6 July 2023. Retrieved 5 July 2023.] Its main uses are relief of congestion due to colds, allergies, and allergic rhinitis.[“Benzedrex”. dailymed.nlm.nih.gov. Archived from the original on 15 July 2023. Retrieved 15 July 2023.] Propylhexedrine is found in over-the-counter inhalers such as Benzedrex. Benzedrex was first manufactured by Smith, Kline and French as an eventual replacement to their earlier inhaler products.[Schwarez J. “Sniffing Benzedrine Inhalers”. Office for Science and Society. McGill University. Archived from the original on 16 July 2023. Retrieved 15 July 2023.] Propylhexedrine is a component in the anticonvulsant preparation barbexaclone. Its S-isomer (levopropylhexedrine or L-propylhexedrine) is bonded with phenobarbital for the purpose of offsetting the barbiturate-induced sedation.[Wesson DR (June 1986). “Propylhexdrine”. Drug and Alcohol Dependence. 17 (2–3): 273–278. doi:10.1016/0376-8716(86)90013-X. PMID 2874970.] Barbexaclone has been known under the brand name of Maliasin, manufactured by Abbott Laboratories, as early as 1965.[Krueger HJ, Schwarz H (April 1965). “[Clinical Communication on the Therapy of Epilepsy With Maliasin]”. Die Medizinische Welt. 14: 690–692. PMID 14276849.][“MALIASIN Trademark – Registration Number 0797076 – Serial Number 72213021”. Justia. Archived from the original on 16 July 2023. Retrieved 16 July 2023.] In 2010, Abbott discontinued sale of its barbexaclone preparation in many countries.[Serracchiani D (31 January 2011). “Parliamentary question | Maliasin | P-001035/2011 | European Parliament”. European Parliament. Archived from the original on 16 July 2023. Retrieved 16 July 2023.] Levopropylhexedrine has been used as an anorectic, under the brand name Eventin.[“Eventin”. Drugs.com. Retrieved 27 March 2013.] Eventin’s use has been documented as early as 1958.[Hofer R, Locker A (April 1958). “[Therapeutic experiences with the appetite depressant eventin]”. Wiener Medizinische Wochenschrift. 108 (14): 304–306. PMID 13558159] Benzedrex is currently manufactured by B.F. Ascher & Co. Inc. Pharmaceuticals.[“Benzedrex Inhaler”. B. F. Ascher and Company. Archived from the original on 8 July 2023. Retrieved 8 July 2023.]
- Propylhexedrine is used to treat acute nasal congestion related to common cold, allergies and hay fever. Use is not to exceed three days.[“Benzedrex”. dailymed.nlm.nih.gov. Archived from the original on 15 July 2023. Retrieved 15 July 2023.] Historically, it has also been used for weight loss in oral tablet preparations at doses ranging from 5 to 30 milligrams.[6][“Obesin Dosage, Interactions”. Archived from the original on 8 July 2023. Retrieved 23 October 2017.] No medications containing propylhexedrine are currently approved for weight loss in the United States.[“NCATS Inxight Drugs — PROPYLHEXEDRINE HYDROCHLORIDE”. drugs.ncats.io. Archived from the original on 16 July 2023. Retrieved 15 July 2023.] Such products have been used in European countries, however, under the trade name Obesin.[Wesson DR (June 1986). “Propylhexdrine”. Drug and Alcohol Dependence. 17 (2–3): 273–278. doi:10.1016/0376-8716(86)90013-X. PMID 2874970.] Obesin has been referenced in literature dating back to the 1950s.[Polster H (February 1965). “[On Poisoning With the Appetite Depressant Propylhexedrine, “Obesin” in a 3-Year-Old Child]”. Archiv für Toxikologie. 20: 271–273. doi:10.1007/BF00577551. PMID 14272412. S2CID 19812917.][Rose W (September 1959). “Arzneimittelsucht durch Mißbrauch von sogenannten Appetitzüglern (Obesin)”. Archiv für Toxikologie (in German). 17 (5): 331–335. doi:10.1007/BF00577633. ISSN 1432-0738. S2CID 31539217.]
- Propylhexedrine should not be used if a MAOI has been used in the past 14 days or is currently in use, for this can result in hypertensive crisis. People with cardiovascular disease should not use propylhexedrine.[“Propylhexedrine Contraindications”. Medscape. Archived from the original on 28 February 2013. Retrieved 8 July 2023.]
- Additionally, stimulant drugs and sympathomimetics should not be taken with propylhexedrine. This can lead to a potentially dangerous spike in blood pressure and irregular heart rhythms.[“Safety of mixing stimulants with medications”. NIDA. Archived from the original on 14 July 2016. Retrieved 8 July 2023.]
- Propylhexedrine is contraindicated in individuals under six years old.[ “Propylhexedrine”. www.mskcc.org. Memorial Sloan Kettering Cancer Center. Retrieved 16 July 2023.] There is at least one case of reported accidental poisoning resulting from a child’s access to a propylhexedrine product.[Polster H (February 1965). “[On Poisoning With the Appetite Depressant Propylhexedrine, “Obesin” in a 3-Year-Old Child]”. Archiv für Toxikologie. 20: 271–273. doi:10.1007/BF00577551. PMID 14272412. S2CID 19812917.]
- As noted by the FDA, the most common symptoms of propylhexedrine overdose are the following; “…[R]apid heart rate, agitation, high blood pressure, chest pain, tremor, hallucinations, delusions, confusion, nausea, and vomiting.”[Center for Drug Evaluation and Research (15 April 2021). “FDA warns that abuse and misuse of the nasal decongestant propylhexedrine causes serious harm”. FDA. U.S. Food and Drug Administration. Archived from the original on 23 February 2023. Retrieved 8 July 2023.] The use of propylhexedrine products in manners inconsistent with their labeling has proven fatal in some cases.[ Sturner WQ, Spruill FG, Garriott JC (July 1974). “Two propylhexedrine-associated fatalities: Benzedrine revisited”. Journal of Forensic Sciences. 19 (3): 572–574. doi:10.1520/JFS10213J. PMID 4137337.]
- As referred to earlier, most of propylhexedrine’s interactions with other medications have to do with its adrenergic properties. This means that propylhexedrine may interact adversely with certain stimulants, bronchodilators, sympathomimetics, nasal decongestants, and antidepressants. Caution should be exercised when administering propylhexedrine concurrently with other medicines.[“Propylhexedrine”. DrugBank. Archived from the original on 15 March 2012. Retrieved 8 July 2023.]
- Propylhexedrine is a TAAR1 agonist. Consequently, it reverses the transporters for dopamine, norepinephrine, and serotonin. This leads to a release of monoamines from presynaptic vesicles into the synaptic cleft. The increased level of monoamines within the synapse results in increased activity at their respective receptors. Additionally, propylhexedrine appears to inhibit VMAT2; resultantly, leading to a further increase in the aforementioned monoamines. Propylhexedrine is also an adrenergic agonist. The pharmacological actions of propylhexedrine are akin to that of structurally similar compounds.[“Propylhexedrine”. DrugBank. Archived from the original on 15 March 2012. Retrieved 8 July 2023.]
- Propylhexedrine undergoes metabolism to form various metabolites including norpropylhexedrine, cyclohexylacetoxime, cis- and trans-4-hydroxypropylhexedrine.[Midha KK, Beckett AH, Saunders A (October 1974). “Identification of the major metabolites of propylhexedrine in vivo (in man) and in vitro (in guinea pig and rabbit)”. Xenobiotica; the Fate of Foreign Compounds in Biological Systems. 4 (10): 627–635. doi:10.1080/00498257409169765. PMID 4428789.]
- Freebase propylhexedrine is a volatile, oily liquid at room temperature. The slow evaporation of freebase propylhexedrine allows it to be administered via inhalation.[US granted 4095596, Grayson M, “Nasal Inhaler”, issued 20 June 1978, assigned to Smithkline Corp.] As an amine, it can easily be protonated to form various salts. These salts include propylhexedrine hydrochloride, propylhexedrine citrate, or propylhexedrine acetate. The salt formed is dependent on the acid used. These salts are stable, clear to off-white crystalline substances that readily dissolve in water.[Mancusi-Ungaro HR, Decker WJ, Forshan VR, Blackwell SJ, Lewis SR (1983). “Tissue injuries associated with parenteral propylhexedrine abuse”. Journal of Toxicology. Clinical Toxicology. 21 (3): 359–372. doi:10.1097/00005373-198307000-00114. PMID 6144800.] Propylhexedrine is structurally similar to phenylethylamines. The only structural difference is the substitution of an alicyclic cyclohexyl group for the aromatic phenyl group of phenethylamine. Propylhexedrine is not an amphetamine, nor even a phenethylamine. It is instead an alkylamine.[“Medical Definition of ALKYLAMINE”. www.merriam-webster.com. Archived from the original on 16 July 2023. Retrieved 15 July 2023.] Propylhexedrine is a chiral compound (the α-carbon is chiral), and the active ingredient contained in Benzedrex inhalers is racemic (RS)-propylhexedrine as the free base.[“Benzedrex Inhaler”. B. F. Ascher and Company. Archived from the original on 8 July 2023. Retrieved 8 July 2023.] (S)-Propylhexedrine, also known as levopropylhexedrine, is believed to be the more biologically active isomer of the two.[Lands AM, Nash VL (April 1947). “The pharmacologic activity of N-methyl-beta-cyclohexyl-isopropylamine hydrochloride”. The Journal of Pharmacology and Experimental Therapeutics. 89 (4): 382–385. PMID 20295519.] The dextrorotatory counterpart, which is mainly unused, is dextropropylhexedrine. (S)-Propylhexedrine can be synthesized from methamphetamine.[Wilson CO, Gisvold O, Doerge RF (1971). Textbook of organic medicinal and pharmaceutical chemistry. Lippincott. p. 491. ISBN 9780397520558.]
- Propylhexedrine can be synthesized starting with cyclohexylacetone in a similar fashion to the phenylacetone synthesis of methamphetamine.[Lednicer D, Mitscher LA (1977). Organic Chemistry of Drug Synthesis. Vol. 1. New York, NY: Wiley. p. 37. ISBN 978-0-471-52141-9.] However, more commonly propylhexedrine is prepared by reacting methamphetamine with Adams’ catalyst. This reduces methamphetamine’s aromatic ring to a cyclohexyl moiety.
- Due to its structure, administration of propylhexedrine can lead to false-positives for phenethylamine-derivatives on urinalysis panels.[Thurman EM, Pedersen MJ, Stout RL, Martin T (1992). “Distinguishing sympathomimetic amines from amphetamine and methamphetamine in urine by gas chromatography/mass spectrometry”. Journal of Analytical Toxicology. 16 (1): 19–27. doi:10.1093/jat/16.1.19. PMID 1640694.]
- Propylhexedrine’s medical use as a decongestant evolved from desires to find safer alternatives to previous agents.[Schwarez J. “Sniffing Benzedrine Inhalers”. Office for Science and Society. McGill University. Archived from the original on 16 July 2023. Retrieved 15 July 2023.] After searching for such an agent, Dr. Glenn E. Ullyot patented propylhexedrine as a decongestant in 1948. This patent was issued for benefit of Smith, Kline & French.[US2454746A, Ullyot, Glenn E., “Cyclohexylalkylamines”, issued 1948-11-23] Subsequently, propylhexedrine underwent market trials in California before being sold nationally in the United States. Propylhexedrine (under the brandname Benzedrex) was first introduced into interstate commerce on August 4, 1949.[Grant G (4 August 1949). “H.R. 2969 – Bezedrine Inhalers”. Congressional Record: Proceedings and Debates of the … Congress (PDF). U.S. Congress. Vol. 95. Government Publishing Office. pp. A5052. Retrieved 15 July 2023.] Approval for use in Canada was granted in 1998.[“Propylhexedrine – NAPRA”. www.napra.ca. Archived from the original on 15 July 2023. Retrieved 15 July 2023.] Barbexaclone, an anticonvulsant containing propylhexedrine, was used in Turkey until its withdrawal from the market in 2009. Barbexaclone’s former niche in Turkish medicine is now largely-occupied by Levetiracetam.[Bolukbasi F, Delil S, Bulus E, Senturk A, Yeni N, Karaagac N (September 2013). “End of the barbexaclone era: an experience of treatment withdrawal”. Epileptic Disorders. 15 (3): 311–313. doi:10.1684/epd.2013.0605. PMID 23981808. S2CID 39112148.]
- The manufacture of propylhexedrine products is typically performed based on guidelines established in government regulations and pharmacopeial monographs.[ “Federal Register Vol. 41, No. 176” (PDF). Government Publishing Office. 9 September 1976. p. 38402.][“USP Monographs: Propylhexedrine Inhalant”. www.pharmacopeia.cn. Archived from the original on 16 July 2023. Retrieved 16 July 2023.] Propylhexedrine was placed under international control by the Convention on Psychotropic Substances in 1985. This action was reversed in 1991.[ “Expert Committee on Drug Dependence’s Twenty-Seventh Report” (PDF). World Health Organization. Expert Committee on Drug Dependence. 28 September 1990. Retrieved 3 July 2023.] On the 4th of April 1988, propylhexedrine was designated a controlled substance (Schedule V) in the United States.[“Federal Register Vol. 53 No. 64” (PDF). National Archives. Archived (PDF) from the original on 28 December 2022. Retrieved 9 July 2022.] This was done to satisfy U.S. compliance with an international treaty. However, in 1991 this action was reversed and propylhexedrine was removed from control under the Controlled Substances Act. This was based on the opinion of the Drug Enforcement Administration that propylhexedrine did not warrant control.[“Federal Register Vol. 56 No. 232” (PDF). National Archives. Archived (PDF) from the original on 28 December 2022. Retrieved 9 July 2022.] The substance has remained unregulated under the Controlled Substances Act in the United States ever since. Furthermore, pursuant to 21 C.F.R 1308.22, Benzedrex is specifically exempt from the Controlled Substances Act.[“Sec. 1308.22 Excluded substances”. Code of Federal Regulations. Drug Enforcement Administration. Archived from the original on 30 May 2023. Retrieved 30 May 2023.][Haislip G (11 January 1989). “Excluded Nonnarcotic Over-the- Counter Substances” (PDF). National Archives and Records Administration. DEA. Archived from the original (PDF) on 16 June 2023. Retrieved 26 June 2023.]
- First reported in the literature as early as 1959, propylhexedrine products have been misused for their monoamine-releasing effects.[Rose W (September 1959). “Arzneimittelsucht durch Mißbrauch von sogenannten Appetitzüglern (Obesin)”. Archiv für Toxikologie (in German). 17 (5): 331–335. doi:10.1007/BF00577633. ISSN 1432-0738. S2CID 31539217.] Recreational misuse can be fatal, its risks are magnified when administering the substance through injection means, and the adverse effects of misuse are similar to that of related substances.[Center for Drug Evaluation and Research (15 April 2021). “FDA warns that abuse and misuse of the nasal decongestant propylhexedrine causes serious harm”. FDA. U.S. Food and Drug Administration. Archived from the original on 23 February 2023. Retrieved 8 July 2023.][Fornazzari L, Carlen PL, Kapur BM (November 1986). “Intravenous abuse of propylhexedrine (Benzedrex) and the risk of brainstem dysfunction in young adults”. The Canadian Journal of Neurological Sciences. Le Journal Canadien des Sciences Neurologiques. 13 (4): 337–339. doi:10.1017/S0317167100036696. PMID 2877725.][“Propylhexedrine (hydrochloride)” (PDF). Safety Data Sheet. Cayman Chemical. Archived from the original (PDF) on 16 April 2014. Retrieved 8 July 2023.][Holler JM, Vorce SP, McDonough-Bender PC, Magluilo J, Solomon CJ, Levine B (January 2011). “A drug toxicity death involving propylhexedrine and mitragynine”. Journal of Analytical Toxicology. 35 (1): 54–59. doi:10.1093/anatox/35.1.54. PMID 21219704.] The undesirable side effects of propylhexedrine at recreational doses are less tolerable compared to other commonly abused monoaminergic substances; consequently, making it less desirable for recreational use.[ “Propylhexedrine (Benzedrex)”. National Capital Poison Control. Archived from the original on 16 July 2023. Retrieved 16 July 2023.][Anderson ED (May 1970). “Propylhexedrine (Benzedrex) psychosis”. The New Zealand Medical Journal. 71 (456): 302. PMID 5270979. S2CID 30786926.][Sturner WQ, Spruill FG, Garriott JC (July 1974). “Two propylhexedrine-associated fatalities: Benzedrine revisited”. Journal of Forensic Sciences. 19 (3): 572–574. doi:10.1520/JFS10213J. PMID 4137337.] Propylhexedrine is also less potent than comparable recreational substances.[Garriott JC (1975). “Editorial: Propylhexadrine – a new dangerous drug?”. Clinical Toxicology. 8 (6): 665–666. doi:10.3109/15563657508990092. PMID 6189.] Even in areas with prevalent substance use, the use of propylhexedrine was reported as not of significant interest.[Smith DE, Wesson DR, Sees KL, Morgan JP (1 October 1988). “An epidemiological and clinical analysis of propylhexedrine abuse in the United States”. Journal of Psychoactive Drugs. 20 (4): 441–442. doi:10.1080/02791072.1988.10472514. PMID 2907528.] The misuse of nasal decongestant,[Anderson ED (May 1970). “Propylhexedrine (Benzedrex) psychosis”. The New Zealand Medical Journal. 71 (456): 302. PMID 5270979. S2CID 30786926.] anoretic,[Rose W (September 1959). “Arzneimittelsucht durch Mißbrauch von sogenannten Appetitzüglern (Obesin)”. Archiv für Toxikologie (in German). 17 (5): 331–335. doi:10.1007/BF00577633. ISSN 1432-0738. S2CID 31539217.] and anticonvulsant preparations [Darcın AE, Dilbaz N, Okay IT (October 2010). “Barbexaclone abuse in a cannabis ex-user”. Substance Abuse. 31 (4): 270–272. doi:10.1080/08897077.2010.514246. PMID 21038181. S2CID 7076881.] have all been reported.
Coenzyme Q – cytochrome c reductase – Oxygen free radicals
A small fraction of electrons leave the electron transport chain before reaching complex IV. Premature electron leakage to oxygen results in the formation of superoxide. The relevance of this otherwise minor side reaction is that superoxide and other reactive oxygen species are highly toxic and are thought to play a role in several pathologies, as well as aging (the free radical theory of aging).[Muller, F. L.; Lustgarten, M. S.; Jang, Y.; Richardson, A. & Van Remmen, H. (2007). “Trends in oxidative aging theories”. Free Radic. Biol. Med. 43 (4): 477–503. doi:10.1016/j.freeradbiomed.2007.03.034. PMID 17640558.] Electron leakage occurs mainly at the Qo site and is stimulated by antimycin A. Antimycin A locks the b hemes in the reduced state by preventing their re-oxidation at the Qi site, which, in turn, causes the steady-state concentrations of the Qo semiquinone to rise, the latter species reacting with oxygen to form superoxide. The effect of high membrane potential is thought to have a similar effect.[Skulachev VP (May 1996). “Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants”. Q. Rev. Biophys. 29 (2): 169–202. doi:10.1017/s0033583500005795. PMID 8870073.] Superoxide produced at the Qo site can be released both into the mitochondrial matrix[Muller F (2000). “The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging”. AGE. 23 (4): 227–253. doi:10.1007/s11357-000-0022-9. PMC 3455268. PMID 23604868.][Muller FL, Liu Y, Van Remmen H (November 2004). “Complex III releases superoxide to both sides of the inner mitochondrial membrane”. J. Biol. Chem. 279 (47): 49064–73. doi:10.1074/jbc.M407715200. PMID 15317809.] and into the intermembrane space, where it can then reach the cytosol.[Muller F (2000). “The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging”. AGE. 23 (4): 227–253. doi:10.1007/s11357-000-0022-9. PMC 3455268. PMID 23604868.][Han D, Williams E, Cadenas E (January 2001). “Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space”. Biochem. J. 353 (Pt 2): 411–6. doi:10.1042/0264-6021:3530411. PMC 1221585. PMID 11139407.] This could be explained by the fact that Complex III might produce superoxide as membrane permeable HOO• rather than as membrane impermeable O−.
2.[Muller FL, Liu Y, Van Remmen H (November 2004). “Complex III releases superoxide to both sides of the inner mitochondrial membrane”. J. Biol. Chem. 279 (47): 49064–73. doi:10.1074/jbc.M407715200. PMID 15317809.]
Coenzyme Q – cytochrome c reductase – Human gene names
MT-CYB: mtDNA encoded cytochrome b; mutations associated with exercise intolerance
CYC1: cytochrome c1
CYCS: cytochrome c
UQCRFS1: Rieske iron sulfur protein
UQCRB: Ubiquinone binding protein, mutation linked with mitochondrial complex III deficiency nuclear type 3
UQCRH: hinge protein
UQCRC2: Core 2, mutations linked to mitochondrial complex III deficiency, nuclear type 5
UQCRC1: Core 1
UQCR: 6.4KD subunit
UQCR10: 7.2KD subunit
TTC19: Newly identified subunit, mutations linked to complex III deficiency nuclear type 2.
Coenzyme Q – cytochrome c reductase – Mutations in complex III genes in human disease
Mutations in complex III-related genes typically manifest as exercise intolerance.[DiMauro S (November 2006). “Mitochondrial myopathies” (PDF). Curr Opin Rheumatol. 18 (6): 636–41. doi:10.1097/01.bor.0000245729.17759.f2. PMID 17053512. S2CID 29140366.][DiMauro S (June 2007). “Mitochondrial DNA medicine”. Biosci. Rep. 27 (1–3): 5–9. doi:10.1007/s10540-007-9032-5. PMID 17484047. S2CID 5849380.] Other mutations have been reported to cause septo-optic dysplasia[Schuelke M, Krude H, Finckh B, Mayatepek E, Janssen A, Schmelz M, Trefz F, Trijbels F, Smeitink J (March 2002). “Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation”. Ann. Neurol. 51 (3): 388–92. doi:10.1002/ana.10151. PMID 11891837. S2CID 12425236.] and multisystem disorders.[Wibrand F, Ravn K, Schwartz M, Rosenberg T, Horn N, Vissing J (October 2001). “Multisystem disorder associated with a missense mutation in the mitochondrial cytochrome b gene”. Ann. Neurol. 50 (4): 540–3. doi:10.1002/ana.1224. PMID 11601507. S2CID 8944744.] However, mutations in BCS1L, a gene responsible for proper maturation of complex III, can result in Björnstad syndrome and the GRACILE syndrome, which in neonates are lethal conditions that have multisystem and neurologic manifestations typifying severe mitochondrial disorders. The pathogenicity of several mutations has been verified in model systems such as yeast.[Fisher N, Castleden CK, Bourges I, Brasseur G, Dujardin G, Meunier B (March 2004). “Human disease-related mutations in cytochrome b studied in yeast”. J. Biol. Chem. 279 (13): 12951–8. doi:10.1074/jbc.M313866200. PMID 14718526.] The extent to which these various pathologies are due to bioenergetic deficits or overproduction of superoxide is presently unknown.
- Mitochondrial chaperone BCS1 (BCS1L), also known as BCS1 homolog, ubiquinol-cytochrome c reductase complex chaperone (h-BCS1), is a protein that in humans is encoded by the BCS1Lgene. BCS1L is a chaperone protein involved in the assembly of Ubiquinol Cytochrome c Reductase (complex III), which is located in the inner mitochondrial membrane and is part of the electron transport chain. Mutations in this gene are associated with mitochondrial complex III deficiency (nuclear, 1), GRACILE syndrome, and Bjoernstad syndrome.[“BCS1L BCS1 homolog, ubiquinol-cytochrome c reductase complex chaperone [Homo sapiens (human)] – Gene – NCBI”. www.ncbi.nlm.nih.gov. Retrieved 2018-08-03.
 This article incorporates text from this source, which is in the public domain.][“BCS1L – Mitochondrial chaperone BCS1 – Homo sapiens (Human) – BCS1L gene & protein”. www.uniprot.org. Retrieved 2018-08-03. This article incorporates text available under the CC BY 4.0][“UniProt: the universal protein knowledgebase”. Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571. PMID 27899622.]
This article incorporates text from this source, which is in the public domain.][“BCS1L – Mitochondrial chaperone BCS1 – Homo sapiens (Human) – BCS1L gene & protein”. www.uniprot.org. Retrieved 2018-08-03. This article incorporates text available under the CC BY 4.0][“UniProt: the universal protein knowledgebase”. Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571. PMID 27899622.] - BCS1L encodes a protein that is located in the inner mitochondrial membrane and involved in the assembly of Ubiquinol Cytochrome c Reductase (complex III). Complex III plays an important role in the mitochondrial respiratory chain by transferring electrons from the Rieskeiron-sulfur protein to cytochrome c. BCS1L is essential for this process through its role in the maintenance of mitochondrial tubular networks, respiratory chain assembly, and formation of the LETM1 complex.[Tamai S, Iida H, Yokota S, Sayano T, Kiguchiya S, Ishihara N, Hayashi J, Mihara K, Oka T (August 2008). “Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L”. Journal of Cell Science. 121 (Pt 15): 2588–600. doi:10.1242/jcs.026625. PMID 18628306.][“BCS1L – Mitochondrial chaperone BCS1 – Homo sapiens (Human) – BCS1L gene & protein”. www.uniprot.org. Retrieved 2018-08-03. This article incorporates text available under the CC BY 4.0][ “UniProt: the universal protein knowledgebase”. Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571. PMID 27899622.]
- Variants of BCS1L have been associated with mitochondrial complex III deficiency, nuclear 1, GRACILE syndrome, and Bjoernstad syndrome. Mitochondrial complex III deficiency, nuclear 1 is a disorder of the mitochondrial respiratory chain resulting in reduced complex III activity and highly variable clinical features usually resulting in multi-system organ failure. Clinical features may include mitochondrial encephalopathy, psychomotor retardation, ataxia, severe failure to thrive, liver dysfunction, renal tubulopathy, muscle weakness, exercise intolerance, lactic acidosis, hypotonia, seizures, and optic atrophy. Pathogenic mutations have included R45C, R56X,[De Meirleir L, Seneca S, Damis E, Sepulchre B, Hoorens A, Gerlo E, et al. (August 2003). “Clinical and diagnostic characteristics of complex III deficiency due to mutations in the BCS1L gene”. American Journal of Medical Genetics. Part A. 121A (2): 126–31. doi:10.1002/ajmg.a.20171. PMID 12910490. S2CID 22246638.][Ramos-Arroyo MA, Hualde J, Ayechu A, De Meirleir L, Seneca S, Nadal N, Briones P (June 2009). “Clinical and biochemical spectrum of mitochondrial complex III deficiency caused by mutations in the BCS1L gene”. Clinical Genetics. 75 (6): 585–7. doi:10.1111/j.1399-0004.2009.01160.x. PMID 19508421. S2CID 205407210.][Gil-Borlado MC, González-Hoyuela M, Blázquez A, García-Silva MT, Gabaldón T, Manzanares J, Vara J, Martín MA, Seneca S, Arenas J, Ugalde C (September 2009). “Pathogenic mutations in the 5′ untranslated region of BCS1L mRNA in mitochondrial complex III deficiency”. Mitochondrion. 9 (5): 299–305. doi:10.1016/j.mito.2009.04.001. PMID 19389488.] T50A,[Blázquez A, Gil-Borlado MC, Morán M, Verdú A, Cazorla-Calleja MR, Martín MA, Arenas J, Ugalde C (February 2009). “Infantile mitochondrial encephalomyopathy with unusual phenotype caused by a novel BCS1L mutation in an isolated complex III-deficient patient”. Neuromuscular Disorders. 19 (2): 143–6. doi:10.1016/j.nmd.2008.11.016. PMID 19162478. S2CID 32624169.] R73C,[Fernandez-Vizarra E, Bugiani M, Goffrini P, Carrara F, Farina L, Procopio E, Donati A, Uziel G, Ferrero I, Zeviani M (May 2007). “Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy”. Human Molecular Genetics. 16 (10): 1241–52. doi:10.1093/hmg/ddm072. PMID 17403714.] P99L, R155P, V353M,[ de Lonlay P, Valnot I, Barrientos A, Gorbatyuk M, Tzagoloff A, Taanman JW, Benayoun E, Chrétien D, Kadhom N, Lombès A, de Baulny HO, Niaudet P, Munnich A, Rustin P, Rötig A (September 2001). “A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure”. Nature Genetics. 29 (1): 57–60. doi:10.1038/ng706. PMID11528392. S2CID10132444] G129R,[Al-Owain M, Colak D, Albakheet A, Al-Younes B, Al-Humaidi Z, Al-Sayed M, et al. (September 2013). “Clinical and biochemical features associated with BCS1L mutation”. Journal of Inherited Metabolic Disease. 36 (5): 813–20. doi:10.1007/s10545-012-9536-4. PMID 22991165. S2CID 13958329.][Tuppen HA, Fehmi J, Czermin B, Goffrini P, Meloni F, Ferrero I, He L, Blakely EL, McFarland R, Horvath R, Turnbull DM, Taylor RW (August 2010). “Long-term survival of neonatal mitochondrial complex III deficiency associated with a novel BCS1L gene mutation”. Molecular Genetics and Metabolism. 100 (4): 345–8. doi:10.1016/j.ymgme.2010.04.010. PMID 20472482.] R183C, F368I,[Fernandez-Vizarra E, Bugiani M, Goffrini P, Carrara F, Farina L, Procopio E, Donati A, Uziel G, Ferrero I, Zeviani M (May 2007). “Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy”. Human Molecular Genetics. 16 (10): 1241–52. doi:10.1093/hmg/ddm072. PMID 17403714.] and S277N. These mutations tend to affect the ATP-binding residues of BCS1L.[Hinson JT, Fantin VR, Schönberger J, Breivik N, Siem G, McDonough B, et al. (February 2007). “Missense mutations in the BCS1L gene as a cause of the Björnstad syndrome”. The New England Journal of Medicine. 356 (8): 809–19. doi:10.1056/NEJMoa055262. PMID 17314340.][ “UniProt: the universal protein knowledgebase”. Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571. PMID 27899622.][“BCS1L – Mitochondrial chaperone BCS1 – Homo sapiens (Human) – BCS1L gene & protein”. www.uniprot.org. Retrieved 2018-08-03. This article incorporates text available under the CC BY 4.0]
- Growth retardation, aminoaciduria, cholestasis, iron overload, lactic acidosis, and early death (GRACILE) is a recessively inherited lethal disease that results in mutli-system organ failure. GRACILE is characterized by fetal growth retardation, lactic acidosis, aminoaciduria, cholestasis, and abnormalities in iron metabolism. Pathogenic mutations have included S78G, R144Q, and V327A.[Visapää I, Fellman V, Vesa J, Dasvarma A, Hutton JL, Kumar V, Payne GS, Makarow M, Van Coster R, Taylor RW, Turnbull DM, Suomalainen A, Peltonen L (October 2002). “GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L”. American Journal of Human Genetics. 71 (4): 863–76. doi:10.1086/342773. PMC 378542. PMID 12215968.][7][“BCS1L – Mitochondrial chaperone BCS1 – Homo sapiens (Human) – BCS1L gene & protein”. www.uniprot.org. Retrieved 2018-08-03. This article incorporates text available under the CC BY 4.0]
- Bjoernstad syndrome is an autosomal recessive disease primarily affecting hearing. This disease is characterized by congenital hearing loss and twisted hairs, a condition known as pili torti, in which hair shafts are flattened at irregular intervals and twisted 180 degrees from the normal axis, making the hair extremely brittle. Pathogenic mutations have included Y301N,[Siddiqi S, Siddiq S, Mansoor A, Oostrik J, Ahmad N, Kazmi SA, Kremer H, Qamar R, Schraders M (December 2013). “Novel mutation in AAA domain of BCS1L causing Bjornstad syndrome”. Journal of Human Genetics. 58 (12): 819–21. doi:10.1038/jhg.2013.101. PMID 24172246.] R184C,[Fernandez-Vizarra E, Bugiani M, Goffrini P, Carrara F, Farina L, Procopio E, Donati A, Uziel G, Ferrero I, Zeviani M (May 2007). “Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy”. Human Molecular Genetics. 16 (10): 1241–52. doi:10.1093/hmg/ddm072. PMID 17403714.] G35R, R114W, R183H, Q302E, and R306H. These mutations tend to affect the protein-protein interactions of BCS1L.[Hinson JT, Fantin VR, Schönberger J, Breivik N, Siem G, McDonough B, et al. (February 2007). “Missense mutations in the BCS1L gene as a cause of the Björnstad syndrome”. The New England Journal of Medicine. 356 (8): 809–19. doi:10.1056/NEJMoa055262. PMID 17314340.][ “UniProt: the universal protein knowledgebase”. Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571. PMID 27899622.][“BCS1L – Mitochondrial chaperone BCS1 – Homo sapiens (Human) – BCS1L gene & protein”. www.uniprot.org. Retrieved 2018-08-03. This article incorporates text available under the CC BY 4.0]
- BCS1L has 11 protein-protein interactions with 8 of them being co-complex interactions. BCS1L has been found to interact with LETM1, DNAJA1, and DDX24.[“14 binary interactions found for search term BCS1L”. IntAct Molecular Interaction Database. EMBL-EBI. Retrieved 2018-08-25.]
- See also
- External links
- Human BCS1L genome location and BCS1L gene details page in the UCSC Genome Browser.
- Further reading
- Maruyama K, Sugano S (January 1994). “Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides”. Gene. 138 (1–2): 171–4. doi:10.1016/0378-1119(94)90802-8. PMID 8125298.
- Fölsch H, Guiard B, Neupert W, Stuart RA (February 1996). “Internal targeting signal of the BCS1 protein: a novel mechanism of import into mitochondria”. The EMBO Journal. 15 (3): 479–87. doi:10.1002/j.1460-2075.1996.tb00380.x. PMC 449966. PMID 8599931.
- Andersson B, Wentland MA, Ricafrente JY, Liu W, Gibbs RA (April 1996). “A “double adaptor” method for improved shotgun library construction”. Analytical Biochemistry. 236 (1): 107–13. doi:10.1006/abio.1996.0138. PMID 8619474.
- Yu W, Andersson B, Worley KC, Muzny DM, Ding Y, Liu W, Ricafrente JY, Wentland MA, Lennon G, Gibbs RA (April 1997). “Large-scale concatenation cDNA sequencing”. Genome Research. 7 (4): 353–8. doi:10.1101/gr.7.4.353. PMC 139146. PMID 9110174.
- Suzuki Y, Yoshitomo-Nakagawa K, Maruyama K, Suyama A, Sugano S (October 1997). “Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library”. Gene. 200 (1–2): 149–56. doi:10.1016/S0378-1119(97)00411-3. PMID 9373149.
- Lubianca Neto JF, Lu L, Eavey RD, Flores MA, Caldera RM, Sangwatanaroj S, Schott JJ, McDonough B, Santos JI, Seidman CE, Seidman JG (May 1998). “The Bjornstad syndrome (sensorineural hearing loss and pili torti) disease gene maps to chromosome 2q34-36”. American Journal of Human Genetics. 62 (5): 1107–12. doi:10.1086/301837. PMC 1377094. PMID 9545407.
- Visapää I, Fellman V, Varilo T, Palotie A, Raivio KO, Peltonen L (November 1998). “Assignment of the locus for a new lethal neonatal metabolic syndrome to 2q33-37”. American Journal of Human Genetics. 63 (5): 1396–403. doi:10.1086/302123. PMC 1377549. PMID 9792866.
- Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, Ishii S, Sugiyama T, Saito K, Isono Y, Irie R, Kushida N, Yoneyama T, Otsuka R, Kanda K, Yokoi T, Kondo H, Wagatsuma M, Murakawa K, Ishida S, Ishibashi T, Takahashi-Fujii A, Tanase T, Nagai K, Kikuchi H, Nakai K, Isogai T, Sugano S (January 2006). “Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes”. Genome Research. 16 (1): 55–65. doi:10.1101/gr.4039406. PMC 1356129. PMID 16344560.
See also
Additional images
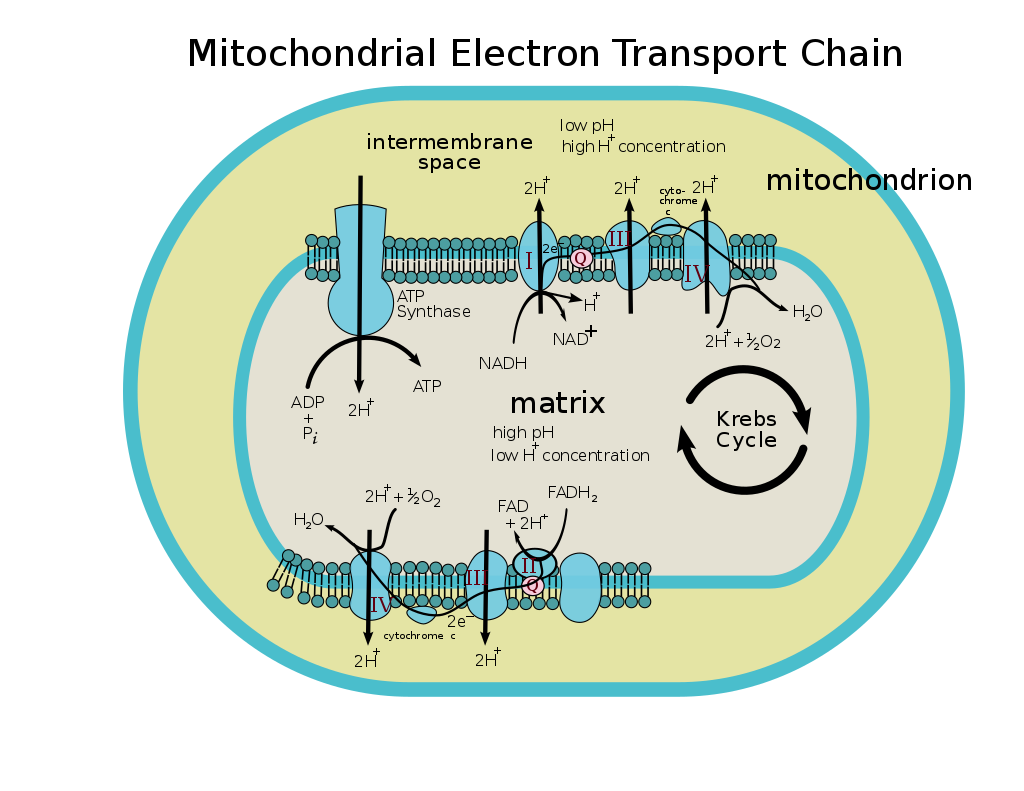
Coenzyme Q – cytochrome c reductase References
- PDB: 1ntz; Gao X, Wen X, Esser L, Quinn B, Yu L, Yu CA, Xia D (August 2003). “Structural basis for the quinone reduction in the bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site”. Biochemistry. 42 (30): 9067–80. doi:10.1021/bi0341814. PMID 12885240.
- Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK (July 1998). “Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex”. Science. 281 (5373): 64–71. Bibcode:1998Sci…281…64I. doi:10.1126/science.281.5373.64. PMID 9651245.
- Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, et al. (1998). “Electron transfer by domain movement in cytochrome bc1”. Nature. 392 (6677): 677–84. Bibcode:1998Natur.392..677Z. doi:10.1038/33612. PMID 9565029. S2CID 4380033.
- Hao GF, Wang F, Li H, Zhu XL, Yang WC, Huang LS, et al. (2012). “Computational discovery of picomolar Q(o) site inhibitors of cytochrome bc1 complex”. J Am Chem Soc. 134 (27): 11168–76. doi:10.1021/ja3001908. PMID 22690928.
- Yang XH, Trumpower BL (1986). “Purification of a three-subunit ubiquinol-cytochrome c oxidoreductase complex from Paracoccus denitrificans”. J Biol Chem. 261 (26): 12282–9. doi:10.1016/S0021-9258(18)67236-9. PMID 3017970.
- Kramer DM, Roberts AG, Muller F, Cape J, Bowman MK (2004). “Q-cycle bypass reactions at the Qo site of the cytochrome bc1 (and related) complexes”. Quinones and Quinone Enzymes, Part B. Meth. Enzymol. Methods in Enzymology. Vol. 382. pp. 21–45. doi:10.1016/S0076-6879(04)82002-0. ISBN 978-0-12-182786-1. PMID 15047094.
- Crofts AR (2004). “The cytochrome bc1 complex: function in the context of structure”. Annu. Rev. Physiol. 66: 689–733. doi:10.1146/annurev.physiol.66.032102.150251. PMID 14977419.
- Ferguson SJ, Nicholls D, Ferguson S (2002). Bioenergetics (3rd ed.). San Diego: Academic. pp. 114–117. ISBN 978-0-12-518121-1.
- Holmes, J. H.; Sapeika, N; Zwarenstein, H (1975). “Inhibitory effect of anti-obesity drugs on NADH dehydrogenase of mouse heart homogenates”. Research Communications in Chemical Pathology and Pharmacology. 11 (4): 645–6. PMID 241101.
- Muller, F. L.; Lustgarten, M. S.; Jang, Y.; Richardson, A. & Van Remmen, H. (2007). “Trends in oxidative aging theories”. Free Radic. Biol. Med. 43 (4): 477–503. doi:10.1016/j.freeradbiomed.2007.03.034. PMID 17640558.
- Skulachev VP (May 1996). “Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants”. Q. Rev. Biophys. 29 (2): 169–202. doi:10.1017/s0033583500005795. PMID 8870073.
- Muller F (2000). “The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging”. AGE. 23 (4): 227–253. doi:10.1007/s11357-000-0022-9. PMC 3455268. PMID 23604868.
- Muller FL, Liu Y, Van Remmen H (November 2004). “Complex III releases superoxide to both sides of the inner mitochondrial membrane”. J. Biol. Chem. 279 (47): 49064–73. doi:10.1074/jbc.M407715200. PMID 15317809.
- Han D, Williams E, Cadenas E (January 2001). “Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space”. Biochem. J. 353 (Pt 2): 411–6. doi:10.1042/0264-6021:3530411. PMC 1221585. PMID 11139407.
- DiMauro S (November 2006). “Mitochondrial myopathies” (PDF). Curr Opin Rheumatol. 18 (6): 636–41. doi:10.1097/01.bor.0000245729.17759.f2. PMID 17053512. S2CID 29140366.
- DiMauro S (June 2007). “Mitochondrial DNA medicine”. Biosci. Rep. 27 (1–3): 5–9. doi:10.1007/s10540-007-9032-5. PMID 17484047. S2CID 5849380.
- Schuelke M, Krude H, Finckh B, Mayatepek E, Janssen A, Schmelz M, Trefz F, Trijbels F, Smeitink J (March 2002). “Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation”. Ann. Neurol. 51 (3): 388–92. doi:10.1002/ana.10151. PMID 11891837. S2CID 12425236.
- Wibrand F, Ravn K, Schwartz M, Rosenberg T, Horn N, Vissing J (October 2001). “Multisystem disorder associated with a missense mutation in the mitochondrial cytochrome b gene”. Ann. Neurol. 50 (4): 540–3. doi:10.1002/ana.1224. PMID 11601507. S2CID 8944744.
- Fisher N, Castleden CK, Bourges I, Brasseur G, Dujardin G, Meunier B (March 2004). “Human disease-related mutations in cytochrome b studied in yeast”. J. Biol. Chem. 279 (13): 12951–8. doi:10.1074/jbc.M313866200. PMID 14718526.
Coenzyme Q – cytochrome c reductase Further reading
- Marres CM, Slater EC (1977). “Polypeptide composition of purified QH2:cytochrome c oxidoreductase from beef-heart mitochondria”. Biochim. Biophys. Acta. 462 (3): 531–548. doi:10.1016/0005-2728(77)90099-8. PMID 597492.
- Rieske JS (1976). “Composition, structure, and function of complex III of the respiratory chain”. Biochim. Biophys. Acta. 456 (2): 195–247. doi:10.1016/0304-4173(76)90012-4. PMID 788795.
- Wikstrom M, Krab K, Saraste M (1981). “Proton-translocating cytochrome complexes”. Annu. Rev. Biochem. 50: 623–655. doi:10.1146/annurev.bi.50.070181.003203. PMID 6267990.
External links
- cytochrome bc1 complex site (Edward A. Berry) at the Wayback Machine (archived October 9, 2006) at lbl.gov
- cytochrome bc1 complex site (Antony R. Crofts) at uiuc.edu
- PROMISE Database: cytochrome bc1 complex at archive.today (archived August 27, 1999) at scripps.edu
- Interactive Molecular Model of Complex III at the Wayback Machine (archived January 12, 2009) (Requires MDL Chime)
- UMich Orientation of Proteins in Membranes families/superfamily-3 – Calculated positions of bc1 and related complexes in membranes
- Coenzyme+Q-Cytochrome-c+Reductase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
| Ion pump: proton pumps: Oxidoreduction-driven transporters (TC 3D) |
|---|
| Mitochondrial proteins |
|---|
| Oxidoreductases: diphenol family (EC 1.10) |
|---|
{kind=link}
Coenzyme Q – cytochrome c reductase Categories:
- EC 7.1.1
- Enzymes of known structure
- Cellular respiration
- Iron–sulfur proteins
- Transmembrane proteins
Ferrochelatase Structure

Human ferrochelatase is a homodimer composed of two 359 amino acid polypeptide chains. It has a total molecular weight of 85.07 kDa.
Each subunit is composed of five regions: a mitochondrial localization sequence, the N terminal domain, two folded domains, and a C terminal extension. Residues 1–62 form a mitochondrial localization domain that is cleaved in post-translational modification. The folded domains contain a total of 17 α-helices and 8 β-sheets. The C terminal extension contains three of the four cysteine residues (Cys403, Cys406, Cys411) that coordinate the catalytic iron–sulfur cluster (2Fe-2S). The fourth coordinating cysteine resides in the N-terminal domain (Cys196).
- Wu, Chia-Kuei; Dailey, Harry A.; Rose, John P.; Burden, Amy; Sellers, Vera M.; Wang, Bi-Cheng (1 February 2001). “The 2.0 Å structure of human ferrochelatase, the terminal enzyme of heme biosynthesis”. Nature Structural Biology. 8 (2): 156–160. doi:10.1038/84152. PMID 11175906. S2CID 9822420.
The active pocket of ferrocheltase consists of two hydrophobic “lips” and a hydrophilic interior. The hydrophobic lips, consisting of the highly conserved residues 300–311, face the inner mitochondrial membrane and facilitate the passage of the poorly soluble protoporphyrin IX substrate and the heme product via the membrane. The interior of the active site pocket contains a highly conserved acidic surface that facilitates proton extraction from protoporphyrin. Histidine and aspartate residues roughly 20 angstroms from the center of the active site on the mitochondrial matrix side of the enzyme coordinate metal binding.
- Wu, Chia-Kuei; Dailey, Harry A.; Rose, John P.; Burden, Amy; Sellers, Vera M.; Wang, Bi-Cheng (1 February 2001). “The 2.0 Å structure of human ferrochelatase, the terminal enzyme of heme biosynthesis”. Nature Structural Biology. 8 (2): 156–160. doi:10.1038/84152. PMID 11175906. S2CID 9822420.
Mechanism

The mechanism of human protoporphyrin metalation remains under investigation. Many researchers have hypothesized distortion of the porphyrin macrocycle as key to catalysis. Researchers studying Bacillus subtilis ferrochelatase propose a mechanism for iron insertion into protoporphyrin in which the enzyme tightly grips rings B, C, and D while bending ring A 36o. Normally planar, this distortion exposes the lone pair of electrons on the nitrogen in ring A to the Fe+2 ion.
- Lecerof, D.; Fodje, M.; Hansson, A.; Hansson, M.; Al-Karadaghi, S. (March 2000). “Structural and mechanistic basis of porphyrin metallation by ferrochelatase”. Journal of Molecular Biology. 297 (1): 221–232. doi:10.1006/jmbi.2000.3569. PMID 10704318.
Subsequent investigation revealed a 100o distortion in protoporphyrin bound to human ferrochelatase. A highly conserved histidine residue (His183 in B. subtilis, His263 in humans) is essential for determining the type of distortion, as well as acting as the initial proton acceptor from protoporphyrin. Anionic residues form a pathway facilitating proton movement away from the catalytic histidine.
- Wu, Chia-Kuei; Dailey, Harry A.; Rose, John P.; Burden, Amy; Sellers, Vera M.; Wang, Bi-Cheng (1 February 2001). “The 2.0 Å structure of human ferrochelatase, the terminal enzyme of heme biosynthesis”. Nature Structural Biology. 8 (2): 156–160. doi:10.1038/84152. PMID 11175906. S2CID 9822420.
- Karlberg, Tobias; Hansson, Mattias D.; Yengo, Raymond K.; Johansson, Renzo; Thorvaldsen, Hege O.; Ferreira, Gloria C.; Hansson, Mats; Al-Karadaghi, Salam (May 2008). “Porphyrin Binding and Distortion and Substrate Specificity in the Ferrochelatase Reaction: The Role of Active Site Residues”. Journal of Molecular Biology. 378 (5): 1074–1083. doi:10.1016/j.jmb.2008.03.040. PMC 2852141. PMID 18423489.
Frataxin Notes
Frataxin is a protein that in humans is encoded by the FXN gene.[Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (Mar 1996). “Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion”. Science. 271 (5254): 1423–7. Bibcode:1996Sci…271.1423C. doi:10.1126/science.271.5254.1423. PMID 8596916. S2CID 20303793.][Carvajal JJ, Pook MA, dos Santos M, Doudney K, Hillermann R, Minogue S, Williamson R, Hsuan JJ, Chamberlain S (Oct 1996). “The Friedreich’s ataxia gene encodes a novel phosphatidylinositol-4- phosphate 5-kinase”. Nature Genetics. 14 (2): 157–62. doi:10.1038/ng1096-157. PMID 8841185. S2CID 6324358.]
It is located in the mitochondrion and Frataxin mRNA is mostly expressed in tissues with a high metabolic rate. The function of frataxin is not clear but it is involved in assembly of iron-sulfur clusters. It has been proposed to act as either an iron chaperone or an iron storage protein. Reduced expression of frataxin is the cause of Friedreich’s ataxia.
Frataxin Structure
X-ray crystallography has shown that human frataxin consists of a β-sheet that supports a pair of parallel α-helices, forming a compact αβ sandwich. Frataxin homologues in other species are similar, sharing the same core structure. However, the frataxin tail sequences, extending from the end of one helix, diverge in sequence and differ in length. Human frataxin has a longer tail sequence than frataxin found in bacteria or yeast. It is hypothesized that the purpose of the tail is to stabilize the protein.[Dhe-Paganon S, Shigeta R, Chi YI, Ristow M, Shoelson SE (Oct 2000). “Crystal structure of human frataxin”. The Journal of Biological Chemistry. 275 (40): 30753–6. doi:10.1074/jbc.C000407200. PMID 10900192.]
Like most mitochondrial proteins, frataxin is synthesized in cytoplasmic ribosomes as large precursor molecules with mitochondrial targeting sequences. Upon entry into mitochondria, the molecules are broken down by a proteolytic reaction to yield mature frataxin.[Stemmler TL, Lesuisse E, Pain, Dancis (August 2010). “Frataxin and Mitochondrial FeS Cluster Biogenesis”. Journal of Biological Chemistry. 285 (35): 26737–26743. doi:10.1074/jbc.R110.118679. PMC 2930671. PMID 20522547.]
Frataxin Function
Frataxin is localized to the mitochondrion. The function of frataxin is not entirely clear, but it seems to be involved in assembly of iron-sulfur clusters. It has been proposed to act as either an iron chaperone or an iron storage protein.[Adinolfi S, Iannuzzi C, Prischi F, Pastore C, Iametti S, Martin SR, Bonomi F, Pastore A (Apr 2009). “Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS”. Nature Structural & Molecular Biology. 16 (4): 390 6. doi:10.1038/nsmb.1579. PMID 19305405. S2CID 205522816.]
Frataxin mRNA is predominantly expressed in tissues with a high metabolic rate (including liver, kidney, brown fat and heart). Mouse and yeast frataxin homologues contain a potential N-terminal mitochondrial targeting sequence, and human frataxin has been observed to co-localise with a mitochondrial protein. Furthermore, disruption of the yeast gene has been shown to result in mitochondrial dysfunction. Friedreich’s ataxia is thus believed to be a mitochondrial disease caused by a mutation in the nuclear genome (specifically, expansion of an intronic GAA triplet repeat in the FXN gene, which encodes the protein frataxin.).[Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (Mar 1996). “Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion”. Science. 271 (5254): 1423–7. Bibcode:1996Sci…271.1423C. doi:10.1126/science.271.5254.1423. PMID 8596916. S2CID 20303793.][Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (Oct 1996). “Clinical and genetic abnormalities in patients with Friedreich’s ataxia”. The New England Journal of Medicine. 335 (16): 1169–75. doi:10.1056/NEJM199610173351601. PMID 8815938.][Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (Aug 1997). “Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin”. Nature Genetics. 16 (4): 345–51. doi:10.1038/ng0897-345. PMID 9241270. S2CID 5883249.]
Frataxin Clinical significance
Reduced expression of frataxin is the cause of Friedreich’s ataxia (FRDA), a neurodegenerative disease. The reduction in frataxin gene expression may be attributable from either the silencing of transcription of the frataxin gene because of epigenetic modifications in the chromosomal entity[Kim E, Napierala M, Dent SY (Oct 2011). “Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich’s ataxia”. Nucleic Acids Research. 39 (19): 8366–77. doi:10.1093/nar/gkr542. PMC 3201871. PMID 21745819.] or from the inability of splicing the expanded GAA repeats in the first intron of the pre-mRNA as seen in bacteria[Pan X, Ding Y, Shi L (Nov 2009). “The roles of SbcCD and RNaseE in the transcription of GAA x TTC repeats in Escherichia coli”. DNA Repair. 8 (11): 1321–7. doi:10.1016/j.dnarep.2009.08.001. PMID 19733517.] and Human cells[Baralle M, Pastor T, Bussani E, Pagani F (Jul 2008). “Influence of Friedreich ataxia GAA noncoding repeat expansions on pre-mRNA processing”. American Journal of Human Genetics. 83 (1): 77–88. doi:10.1016/j.ajhg.2008.06.018. PMC 2443835. PMID 18597733.] or both. The expansion of intronic trinucleotide repeat GAA results in Friedreich’s ataxia.[“Entrez Gene: FXN frataxin”.] This expanded repeat causes R-loop formation, and using a repeat-targeted oligonucleotide to disrupt the R-loop can reactivate frataxin expression.[Li L, Matsui M, Corey DR (2016-01-01). “Activating frataxin expression by repeat-targeted nucleic acids”. Nature Communications. 7: 10606. Bibcode:2016NatCo…710606L. doi:10.1038/ncomms10606. PMC 4742999. PMID 26842135.]
- Friedreich’s ataxia (FRDA or FA) is an autosomal-recessive genetic disease that causes difficulty walking, a loss of coordination in the arms and legs, and impaired speech that worsens over time. Symptoms generally start between 5 and 20 years of age. Many develop hypertrophic cardiomyopathy and require a mobility aid such as a cane, walker, or wheelchair in their teens. As the disease progresses, some affected people lose their sight and hearing. Other complications may include scoliosis and diabetes mellitus.
- The condition is caused by mutations in the FXN gene on chromosome 9, which makes a protein called frataxin. In FRDA, cells produce less frataxin. Degeneration of nerve tissue in the spinal cord causes the ataxia; particularly affected are the sensory neurons essential for directing muscle movement of the arms and legs through connections with the cerebellum. The spinal cord becomes thinner, and nerve cells lose some myelin sheath.
- In February 2023, the first approval of a treatment for FA was granted by the FDA. Approval in the EU is pending. There are several additional therapies in trial. FRDA shortens life expectancy due to heart disease, but some people can live into their 60s or older. FRDA affects one in 50,000 people in the United States and is the most common inherited ataxia. Rates are highest in people of Western European descent. The condition is named after German physician Nikolaus Friedreich, who first described it in the 1860s.
96% of FRDA patients have a GAA trinucleotide repeat expansion in intron 1 of both alleles of their FXN gene.[Clark E, Johnson J, Dong YN, Mercado-Ayon, Warren N, Zhai M, McMillan E, Salovin A, Lin H, Lynch DR (November 2018). “Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease”. Neuronal Signaling. 2 (4): NS20180060. doi:10.1042/NS20180060. PMC 7373238. PMID 32714592.] Overall, this leads to a decrease in frataxin mRNA synthesis and a decrease (but not absence) in frataxin protein in people with FRDA. (A subset of FRDA patients have GAA expansion in one chromosome and a point mutation in the FXN exon in the other chromosome.) In the typical case, the length of the allele with the shorter GAA expansion inversely correlates with frataxin levels. FRDA patients’ peripheral tissues typically have less than 10% of the frataxin levels exhibited by unaffected people.[Clark E, Johnson J, Dong YN, Mercado-Ayon, Warren N, Zhai M, McMillan E, Salovin A, Lin H, Lynch DR (November 2018). “Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease”. Neuronal Signaling. 2 (4): NS20180060. doi:10.1042/NS20180060. PMC 7373238. PMID 32714592.] Lower levels of frataxin result in earlier disease onset and faster progression.
FRDA is characterized by ataxia, sensory loss, and cardiomyopathy. The reason frataxin deficiency causes these symptoms is not entirely clear. On a cellular level, it is linked to iron accumulation in the mitochondria and increased oxidant sensitivity. For reasons that are not well understood, this primarily affects the tissue of the dorsal root ganglia, cerebellum, and heart muscle.[Stemmler TL, Lesuisse E, Pain, Dancis (August 2010). “Frataxin and Mitochondrial FeS Cluster Biogenesis”. Journal of Biological Chemistry. 285 (35): 26737–26743. doi:10.1074/jbc.R110.118679. PMC 2930671. PMID 20522547.]
Frataxin Animal studies
In mice, complete inactivation of the FXN gene is lethal in the early embryonic stage.[Cossée M, Puccio H, Gansmuller A, Koutnikova H, Dierich A, LeMeur M, Fischbeck K, Dollé P, Kœnig M (May 2000). “Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation”. Human Molecular Genetics. 9 (8): 1219–1226. doi:10.1093/hmg/9.8.1219. PMID 10767347. Archived from the original on 2 June 2018. Retrieved 5 April 2019.] Although nearly all organisms express a frataxin homologue, the GAA repeat in intron 1 only exists in humans and other primates, so the mutation that causes FDRA can’t occur naturally in other animals. Scientists have developed several options to model this disease in mice. One approach is to silence frataxin expression in just one specific tissue type of interest: the heart (mice modified this way are called MCK), all neurons (NSE), or just the spinal cord and cerebellum (PRP).[Perdomini M, Hick A, Puccio H (17 July 2013). “Animal and cellular models of Friedreich ataxia”. Journal of Neurochemistry. 126: 65–79. doi:10.1111/jnc.12219. PMID 23859342. S2CID 1427817.]
Another approach involves inserting a GAA expansion into the first intron of the mouse FXN gene, which should inhibit frataxin production, just like in humans. Mice that are homozygous for this modified gene are called KIKI (knock-in knock-in), and the compound heterozygotes formed by crossing KIKI mice with frataxin knockout mice are called KIKO (knock-in knock-out). However, even KIKO mice still express 25-36% of the normal frataxin level, and show very mild symptoms. The final approach involves creating transgenic mice with a GAA-expanded version of the human frataxin gene. These mice are called YG22R (one GAA sequence of 190 repeats) and YG22R (two GAA sequences of 90 and 190 repeats). These mice show symptoms similar to human patients.[Perdomini M, Hick A, Puccio H (17 July 2013). “Animal and cellular models of Friedreich ataxia”. Journal of Neurochemistry. 126: 65–79. doi:10.1111/jnc.12219. PMID 23859342. S2CID 1427817.]
An overexpression of frataxin in Drosophila has shown an increase in antioxidant capability, resistance to oxidative stress insults and longevity,[Runko AP, Griswold AJ, Min KT (March 2008). “Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila”. FEBS Letters. 582 (5): 715–9. doi:10.1016/j.febslet.2008.01.046. PMID 18258192. S2CID 207603250.] supporting the theory that the role of frataxin is to protect the mitochondria from oxidative stress and the ensuing cellular damage.
Fibroblasts from a mouse model of FRDA and FRDA patient fibroblasts show increased levels of DNA double-strand breaks.[Khonsari H, Schneider M, Al-Mahdawi S, Chianea YG, Themis M, Parris C, Pook MA, Themis M (December 2016). “Lentivirus-meditated frataxin gene delivery reverses genome instability in Friedreich ataxia patient and mouse model fibroblasts”. Gene Ther. 23 (12): 846–856. doi:10.1038/gt.2016.61. PMC 5143368. PMID 27518705.]
A lentivirus gene delivery system was used to deliver the frataxin gene to the FRDA mouse model and human patient cells, and this resulted in long-term restored expression of frataxin mRNA and frataxin protein. This restored expression of the frataxin gene was accompanied by a substantial reduction in the number of DNA double-strand breaks.[Khonsari H, Schneider M, Al-Mahdawi S, Chianea YG, Themis M, Parris C, Pook MA, Themis M (December 2016). “Lentivirus-meditated frataxin gene delivery reverses genome instability in Friedreich ataxia patient and mouse model fibroblasts”. Gene Ther. 23 (12): 846–856. doi:10.1038/gt.2016.61. PMC 5143368. PMID 27518705.]
The impaired frataxin in FRDA cells appears to cause reduced capacity for repair of DNA damage and this may contribute to neurodegeneration.[Khonsari H, Schneider M, Al-Mahdawi S, Chianea YG, Themis M, Parris C, Pook MA, Themis M (December 2016). “Lentivirus-meditated frataxin gene delivery reverses genome instability in Friedreich ataxia patient and mouse model fibroblasts”. Gene Ther. 23 (12): 846–856. doi:10.1038/gt.2016.61. PMC 5143368. PMID 27518705.]
Frataxin Interactions
Frataxin has been shown to biologically interact with the enzyme PMPCB.[Koutnikova H, Campuzano V, Koenig M (Sep 1998). “Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase”. Human Molecular Genetics. 7 (9): 1485–9. doi:10.1093/hmg/7.9.1485. PMID 9700204.]
- Mitochondrial-processing peptidase subunit beta is an enzyme that in humans is encoded by the PMPCBgene.[Mao M, Fu G, Wu JS, Zhang QH, Zhou J, Kan LX, Huang QH, He KL, Gu BW, Han ZG, Shen Y, Gu J, Yu YP, Xu SH, Wang YX, Chen SJ, Chen Z (Jul 1998). “Identification of genes expressed in human CD34(+) hematopoietic stem/progenitor cells by expressed sequence tags and efficient full-length cDNA cloning”. Proceedings of the National Academy of Sciences of the United States of America. 95 (14): 8175–80. Bibcode:1998PNAS…95.8175M. doi:10.1073/pnas.95.14.8175. PMC 20949. PMID 9653160.][“Entrez Gene: PMPCB peptidase (mitochondrial processing) beta”] This gene is a member of the peptidase M16 family and encodes a protein with a zinc-binding motif. This protein is located in the mitochondrial matrix and catalyzes the cleavage of the leader peptides of precursor proteins newly imported into the mitochondria, though it only functions as part of a heterodimeric complex.[“Entrez Gene: PMPCB peptidase (mitochondrial processing) beta”]
- As the beta subunit of Mitochondrial-processing peptidase, PMPCB forms a heterodimer with the subunit Mitochondrial-processing peptidase subunit alpha. In addition, PMPCB has been shown to interact with PMPCA and Frataxin.[8]
- Mitochondrial-processing peptidase subunit alpha is an enzyme that in humans is encoded by the PMPCAgene.[Nagase T, Seki N, Tanaka A, Ishikawa K, Nomura N (Mar 1996). “Prediction of the coding sequences of unidentified human genes. IV. The coding sequences of 40 new genes (KIAA0121-KIAA0160) deduced by analysis of cDNA clones from human cell line KG-1”. DNA Res. 2 (4): 167–74, 199–210. doi:10.1093/dnares/2.4.167. PMID 8590280.][ Nagase T, Miyajima N, Tanaka A, Sazuka T, Seki N, Sato S, Tabata S, Ishikawa K, Kawarabayasi Y, Kotani H, et al. (Jul 1995). “Prediction of the coding sequences of unidentified human genes. III. The coding sequences of 40 new genes (KIAA0081-KIAA0120) deduced by analysis of cDNA clones from human cell line KG-1”. DNA Res. 2 (1): 37–43. doi:10.1093/dnares/2.1.37. PMID 7788527.][“Entrez Gene: PMPCA peptidase (mitochondrial processing) alpha”] This gene PMPCA encoded a protein that is a member of the peptidase M16 family. This protein is located in the mitochondrial matrix and catalyzes the cleavage of the leader peptides of precursor proteins newly imported into the mitochondria, though it only functions as part of a heterodimeric complex.
- Mitochondrial-processing peptidase (MPP) is a metalloendopeptidase, containing two structurally related subunits, Subunit alpha and mitochondrial-processing peptidase subunit beta, working in conjunction for its catalytic function.[Teixeira PF, Glaser E (Feb 2013). “Processing peptidases in mitochondria and chloroplasts”. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 1833 (2): 360–70. doi:10.1016/j.bbamcr.2012.03.012. PMID 22495024.] Containing the catalytic site, the beta subunit PMPCB protein cleaves presequences (transit peptides) from mitochondrial protein precursors and releases of N-terminal transit peptides from precursor proteins imported into the mitochondrion, typically with Arg in position P2.
- As the alpha subunit of Mitochondrial-processing peptidase, PMPCA forms a heterodimer with the subunit PMPCB.
- The majority of mitochondrial proteins is nuclear-coded, which necessitates proper translocations of mitochondrial targeting proteins. Many mitochondrial proteins are synthesized in a precursor form that contains mitochondria targeting sequence. These precursors are usually cleaved by peptidases and proteases before they arrive their sub-organellar locations. It is likely that altered activity of the mitochondrial processing peptidases is essential to ensure the correct maturation of mitochondrial proteins and that altered activity of these proteases will have dramatic effects in the activity, stability and assembly of mitochondrial proteins. Evidences showed that MPP was involved in the proteolytic maturation of Frataxin, a protein responsible for iron homeostasis.[Branda SS, Yang ZY, Chew A, Isaya G (Jun 1999). “Mitochondrial intermediate peptidase and the yeast frataxin homolog together maintain mitochondrial iron homeostasis in Saccharomyces cerevisiae”. Human Molecular Genetics. 8 (6): 1099–110. doi:10.1093/hmg/8.6.1099. PMID 10332043.] Accordingly, MPP deficiency was shown to be involved in Friedreich ataxia, an autossomic recessive neurodegenerative disorder[Cavadini P, Adamec J, Taroni F, Gakh O, Isaya G (Dec 2000). “Two-step processing of human frataxin by mitochondrial processing peptidase. Precursor and intermediate forms are cleaved at different rates”. The Journal of Biological Chemistry. 275 (52): 41469–75.][Patel PI, Isaya G (Jul 2001). “Friedreich ataxia: from GAA triplet-repeat expansion to frataxin deficiency”. American Journal of Human Genetics. 69 (1): 15–24. doi:10.1086/321283. PMC 1226030. PMID 11391483.]
- See Also
- Schwer B, North BJ, Frye RA, Ott M, Verdin E (Aug 2002). “The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase”. The Journal of Cell Biology. 158 (4): 647–57. doi:10.1083/jcb.200205057. PMC 2174009. PMID 12186850.
- Luciano P, Geoffroy S, Brandt A, et al. (1997). “Functional cooperation of the mitochondrial processing peptidase subunits”. J. Mol. Biol. 272 (2): 213–25. doi:10.1006/jmbi.1997.1231. PMID 9299349.
- Nagao Y, Kitada S, Kojima K, et al. (2000). “Glycine-rich region of mitochondrial processing peptidase alpha-subunit is essential for binding and cleavage of the precursor proteins”. J. Biol. Chem. 275 (44): 34552–6. doi:10.1074/jbc.M003110200. PMID 10942759.
- Harris RA, Yang A, Stein RC, et al. (2002). “Cluster analysis of an extensive human breast cancer cell line protein expression map database”. Proteomics. 2 (2): 212–23. doi:10.1002/1615-9861(200202)2:2<212::AID-PROT212>3.0.CO;2-H. PMID 11840567. S2CID 44946014.
- Hassel S, Eichner A, Yakymovych M, et al. (2004). “Proteins associated with type II bone morphogenetic protein receptor (BMPR-II) and identified by two-dimensional gel electrophoresis and mass spectrometry”. Proteomics. 4 (5): 1346–58. doi:10.1002/pmic.200300770. PMID 15188402. S2CID 6773754.
Frataxin References
- GRCh38: Ensembl release 89: ENSG00000165060 – Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000059363 – Ensembl, May 2017
- “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- “Mouse PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (Mar 1996). “Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion”. Science. 271 (5254): 1423–7. Bibcode:1996Sci…271.1423C. doi:10.1126/science.271.5254.1423. PMID 8596916. S2CID 20303793.
- Carvajal JJ, Pook MA, dos Santos M, Doudney K, Hillermann R, Minogue S, Williamson R, Hsuan JJ, Chamberlain S (Oct 1996). “The Friedreich’s ataxia gene encodes a novel phosphatidylinositol-4- phosphate 5-kinase”. Nature Genetics. 14 (2): 157–62. doi:10.1038/ng1096-157. PMID 8841185. S2CID 6324358.
- Dhe-Paganon S, Shigeta R, Chi YI, Ristow M, Shoelson SE (Oct 2000). “Crystal structure of human frataxin”. The Journal of Biological Chemistry. 275 (40): 30753–6. doi:10.1074/jbc.C000407200. PMID 10900192.
- Stemmler TL, Lesuisse E, Pain, Dancis (August 2010). “Frataxin and Mitochondrial FeS Cluster Biogenesis”. Journal of Biological Chemistry. 285 (35): 26737–26743. doi:10.1074/jbc.R110.118679. PMC 2930671. PMID 20522547.
- Adinolfi S, Iannuzzi C, Prischi F, Pastore C, Iametti S, Martin SR, Bonomi F, Pastore A (Apr 2009). “Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS”. Nature Structural & Molecular Biology. 16 (4): 390–6. doi:10.1038/nsmb.1579. PMID 19305405. S2CID 205522816.
- Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (Oct 1996). “Clinical and genetic abnormalities in patients with Friedreich’s ataxia”. The New England Journal of Medicine. 335 (16): 1169–75. doi:10.1056/NEJM199610173351601. PMID 8815938.
- Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (Aug 1997). “Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin”. Nature Genetics. 16 (4): 345–51. doi:10.1038/ng0897-345. PMID 9241270. S2CID 5883249.
- Kim E, Napierala M, Dent SY (Oct 2011). “Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich’s ataxia”. Nucleic Acids Research. 39 (19): 8366–77. doi:10.1093/nar/gkr542. PMC 3201871. PMID 21745819.
- Pan X, Ding Y, Shi L (Nov 2009). “The roles of SbcCD and RNaseE in the transcription of GAA x TTC repeats in Escherichia coli”. DNA Repair. 8 (11): 1321–7. doi:10.1016/j.dnarep.2009.08.001. PMID 19733517.
- Baralle M, Pastor T, Bussani E, Pagani F (Jul 2008). “Influence of Friedreich ataxia GAA noncoding repeat expansions on pre-mRNA processing”. American Journal of Human Genetics. 83 (1): 77–88. doi:10.1016/j.ajhg.2008.06.018. PMC 2443835. PMID 18597733.
- “Entrez Gene: FXN frataxin”.
- Li L, Matsui M, Corey DR (2016-01-01). “Activating frataxin expression by repeat-targeted nucleic acids”. Nature Communications. 7: 10606. Bibcode:2016NatCo…710606L. doi:10.1038/ncomms10606. PMC 4742999. PMID 26842135.
- Clark E, Johnson J, Dong YN, Mercado-Ayon, Warren N, Zhai M, McMillan E, Salovin A, Lin H, Lynch DR (November 2018). “Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease”. Neuronal Signaling. 2 (4): NS20180060. doi:10.1042/NS20180060. PMC 7373238. PMID 32714592.
- Cossée M, Puccio H, Gansmuller A, Koutnikova H, Dierich A, LeMeur M, Fischbeck K, Dollé P, Kœnig M (May 2000). “Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation”. Human Molecular Genetics. 9 (8): 1219–1226. doi:10.1093/hmg/9.8.1219. PMID 10767347. Archived from the original on 2 June 2018. Retrieved 5 April 2019.
- Perdomini M, Hick A, Puccio H (17 July 2013). “Animal and cellular models of Friedreich ataxia”. Journal of Neurochemistry. 126: 65–79. doi:10.1111/jnc.12219. PMID 23859342. S2CID 1427817.
- Runko AP, Griswold AJ, Min KT (March 2008). “Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila”. FEBS Letters. 582 (5): 715–9. doi:10.1016/j.febslet.2008.01.046. PMID 18258192. S2CID 207603250.
- Khonsari H, Schneider M, Al-Mahdawi S, Chianea YG, Themis M, Parris C, Pook MA, Themis M (December 2016). “Lentivirus-meditated frataxin gene delivery reverses genome instability in Friedreich ataxia patient and mouse model fibroblasts”. Gene Ther. 23 (12): 846–856. doi:10.1038/gt.2016.61. PMC 5143368. PMID 27518705.
- Koutnikova H, Campuzano V, Koenig M (Sep 1998). “Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase”. Human Molecular Genetics. 7 (9): 1485–9. doi:10.1093/hmg/7.9.1485. PMID 9700204.
Frataxin Further reading
- Thierbach R, Drewes G, Fusser M, Voigt A, Kuhlow D, Blume U, Schulz TJ, Reiche C, Glatt H, Epe B, Steinberg P, Ristow M (Nov 2010). “The Friedreich’s ataxia protein frataxin modulates DNA base excision repair in prokaryotes and mammals”. The Biochemical Journal. 432 (1): 165–72. doi:10.1042/BJ20101116. PMC 2976068. PMID 20819074.
- Montermini L, Rodius F, Pianese L, Moltò MD, Cossée M, Campuzano V, Cavalcanti F, Monticelli A, Palau F, Gyapay G (Nov 1995). “The Friedreich ataxia critical region spans a 150-kb interval on chromosome 9q13”. American Journal of Human Genetics. 57 (5): 1061–7. PMC 1801369. PMID 7485155.
- Bidichandani SI, Ashizawa T, Patel PI (May 1997). “Atypical Friedreich ataxia caused by compound heterozygosity for a novel missense mutation and the GAA triplet-repeat expansion”. American Journal of Human Genetics. 60 (5): 1251–6. PMC 1712428. PMID 9150176.
- Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J (Jun 1997). “Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin”. Science. 276 (5319): 1709–12. doi:10.1126/science.276.5319.1709. PMID 9180083.
- Koutnikova H, Campuzano V, Foury F, Dollé P, Cazzalini O, Koenig M (Aug 1997). “Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin”. Nature Genetics. 16 (4): 345–51. doi:10.1038/ng0897-345. PMID 9241270. S2CID 5883249.
- Wilson RB, Roof DM (Aug 1997). “Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue”. Nature Genetics. 16 (4): 352–7. doi:10.1038/ng0897-352. PMID 9241271. S2CID 22652291.
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Dürr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M (Oct 1997). “Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes”. Human Molecular Genetics. 6 (11): 1771–80. doi:10.1093/hmg/6.11.1771. PMID 9302253.
- Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (Oct 1997). “Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia”. Nature Genetics. 17 (2): 215–7. doi:10.1038/ng1097-215. PMID 9326946. S2CID 23151137.
- Jiralerspong S, Liu Y, Montermini L, Stifani S, Pandolfo M (1997). “Frataxin shows developmentally regulated tissue-specific expression in the mouse embryo”. Neurobiology of Disease. 4 (2): 103–13. doi:10.1006/nbdi.1997.0139. PMID 9331900. S2CID 6520439.
- Koutnikova H, Campuzano V, Koenig M (Sep 1998). “Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase”. Human Molecular Genetics. 7 (9): 1485–9. doi:10.1093/hmg/7.9.1485. PMID 9700204.
- Zühlke C, Laccone F, Cossée M, Kohlschütter A, Koenig M, Schwinger E (Jul 1998). “Mutation of the start codon in the FRDA1 gene: linkage analysis of three pedigrees with the ATG to ATT transversion points to a unique common ancestor”. Human Genetics. 103 (1): 102–5. doi:10.1007/s004390050791. PMID 9737785. S2CID 26999143.
- Bartolo C, Mendell JR, Prior TW (Oct 1998). “Identification of a missense mutation in a Friedreich’s ataxia patient: implications for diagnosis and carrier studies”. American Journal of Medical Genetics. 79 (5): 396–9. doi:10.1002/(SICI)1096-8628(19981012)79:5<396::AID-AJMG13>3.0.CO;2-M. PMID 9779809.
- Cossée M, Dürr A, Schmitt M, Dahl N, Trouillas P, Allinson P, Kostrzewa M, Nivelon-Chevallier A, Gustavson KH, Kohlschütter A, Müller U, Mandel JL, Brice A, Koenig M, Cavalcanti F, Tammaro A, De Michele G, Filla A, Cocozza S, Labuda M, Montermini L, Poirier J, Pandolfo M (Feb 1999). “Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes”. Annals of Neurology. 45 (2): 200–6. doi:10.1002/1531-8249(199902)45:2<200::AID-ANA10>3.0.CO;2-U. PMID 9989622. S2CID 24885238.
- Coppola G, De Michele G, Cavalcanti F, Pianese L, Perretti A, Santoro L, Vita G, Toscano A, Amboni M, Grimaldi G, Salvatore E, Caruso G, Filla A (May 1999). “Why do some Friedreich’s ataxia patients retain tendon reflexes? A clinical, neurophysiological and molecular study”. Journal of Neurology. 246 (5): 353–7. doi:10.1007/s004150050362. PMID 10399865. S2CID 7367457.
- Branda SS, Cavadini P, Adamec J, Kalousek F, Taroni F, Isaya G (Aug 1999). “Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase”. The Journal of Biological Chemistry. 274 (32): 22763–9. doi:10.1074/jbc.274.32.22763. PMID 10428860.
- Gordon DM, Shi Q, Dancis A, Pain D (Nov 1999). “Maturation of frataxin within mammalian and yeast mitochondria: one-step processing by matrix processing peptidase”. Human Molecular Genetics. 8 (12): 2255–62. doi:10.1093/hmg/8.12.2255. PMID 10545606.
- Forrest SM, Knight M, Delatycki MB, Paris D, Williamson R, King J, Yeung L, Nassif N, Nicholson GA (Aug 1998). “The correlation of clinical phenotype in Friedreich ataxia with the site of point mutations in the FRDA gene”. Neurogenetics. 1 (4): 253–7. doi:10.1007/s100480050037. PMID 10732799. S2CID 7463903.
- Al-Mahdawi S, Pook M, Chamberlain S (Jul 2000). “A novel missense mutation (L198R) in the Friedreich’s ataxia gene”. Human Mutation. 16 (1): 95. doi:10.1002/1098-1004(200007)16:1<95::AID-HUMU29>3.0.CO;2-E. PMID 10874325. S2CID 26295274.
Frataxin External links
- GeneReviews/NCBI/NIH/UW entry on Friedreich Ataxia
- frataxin at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- Overview of all the structural information available in the PDB for UniProt: Q16595 (Frataxin, mitochondrial) at the PDBe-KB.
| Mitochondrial proteins |
|---|
Frataxin Categories:
Frataxin chaperones iron to the matrix side of ferrochelatase, where aspartate and histidine residues on both proteins coordinate iron transfer into ferrochelatase.
- Bencze, Krisztina Z.; Yoon, Taejin; Mill?n-Pacheco, C?sar; Bradley, Patrick B.; Pastor, Nina; Cowan, J. A.; Stemmler, Timothy L. (2007). “Human frataxin: iron and ferrochelatase binding surface”. Chemical Communications (18): 1798–1800. doi:10.1039/B703195E. PMC 2862461. PMID 17476391.
Two arginine and tyrosine residues in the active site (Arg164, Tyr165) may perform the final metalation.
- Wu, Chia-Kuei; Dailey, Harry A.; Rose, John P.; Burden, Amy; Sellers, Vera M.; Wang, Bi-Cheng (1 February 2001). “The 2.0 Å structure of human ferrochelatase, the terminal enzyme of heme biosynthesis”. Nature Structural Biology. 8 (2): 156–160. doi:10.1038/84152. PMID 11175906. S2CID 9822420.

Clinical significance
Defects in ferrochelatase create a buildup of protoporphyrin IX, causing erythropoietic protoporphyria (EPP).
- James, William D.; Berger, Timothy G. (2006). Andrews’ Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
The disease can result from a variety of mutations in FECH, most of which behave in an autosomal dominant manner with low clinical penetrance. Clinically, patients with EPP present with a range of symptoms, from asymptomatic to suffering from an extremely painful photosensitivity. In less than five percent of cases, accumulation of protoporphyrin in the liver results in cholestasis (blockage of bile flow from the liver to the small intestine) and terminal liver failure.
- Rüfenacht, U.B.; Gouya, L.; Schneider-Yin, X.; Puy, H.; Schäfer, B.W.; Aquaron, R.; Nordmann, Y.; Minder, E.I.; Deybach, J.C. (1998). “Systematic Analysis of Molecular Defects in the Ferrochelatase Gene from Patients with Erythropoietic Protoporphyria”. The American Journal of Human Genetics. 62 (6): 1341–52. doi:10.1086/301870. PMC 1377149. PMID 9585598.
In cases of lead poisoning, lead inhibits ferrochelatase activity, in part resulting in porphyria.
- “Lead Toxicity — What Are Possible Health Effects from Lead Exposure?”. Agency for Toxic Substances & Disease Registry. Retrieved 9 February 2021.
Interactions
Ferrochelatase interacts with numerous other enzymes involved in heme biosynthesis, catabolism, and transport, including protoporphyrinogen oxidase, 5-aminolevulinate synthase, ABCB10, ABCB7, succinyl-CoA synthetase, and mitoferrin-1. Multiple studies have suggested the existence of an oligomeric complex that enables substrate channeling and coordination of overall iron and porphyrin metabolism throughout the cell.
- Medlock, Amy E.; Shiferaw, Mesafint T.; Marcero, Jason R.; Vashisht, Ajay A.; Wohlschlegel, James A.; Phillips, John D.; Dailey, Harry A.; Liesa, Marc (19 August 2015). “Identification of the Mitochondrial Heme Metabolism Complex”. PLOS ONE. 10 (8): e0135896. Bibcode:2015PLoSO..1035896M. doi:10.1371/journal.pone.0135896. PMC 4545792. PMID 26287972.
- Chen, W.; Dailey, H. A.; Paw, B. H. (28 April 2010). “Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis”. Blood. 116 (4): 628–630. doi:10.1182/blood-2009-12-259614. PMC 3324294. PMID 20427704.
ATP-binding cassette sub-family B member 7, mitochondrial is a protein that in humans is encoded by the ABCB7gene.[Savary S, Allikmets R, Denizot F, Luciani MF, Mattei MG, Dean M, Chimini G (July 1997). “Isolation and chromosomal mapping of a novel ATP-binding cassette transporter conserved in mouse and human”. Genomics. 41 (2): 275–8. doi:10.1006/geno.1997.4658. PMID 9143506][“Entrez Gene: ABCB7 ATP-binding cassette, sub-family B (MDR/TAP), member 7”.] The membrane-associated protein encoded by this gene is a member of the superfamily of ATP-binding cassette (ABC) transporters. ABC proteins transport various molecules across extra- and intra-cellular membranes. ABC genes are divided into seven distinct subfamilies (ABC1, MDR/TAP, MRP, ALD, OABP, GCN20, White). Forty eight ABC genes have been reported in humans. Among these, many have been characterized and shown to be causally related to diseases present in humans such as cystic fibrosis, adrenoleukodystrophy, Stargardt disease, drug-resistant tumors, Dubin–Johnson syndrome, Byler’s disease, progressive familiar intrahepatic cholestasis, X-linked sideroblastic anemia, ataxia, and persistent and hyperinsulimenic hypoglycemia.[Choi CH (Oct 2005). “ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal”. Cancer Cell International. 5: 30. doi:10.1186/1475-2867-5-30. PMC 1277830. PMID 16202168.] ABC transporters are also involved in multiple drug resistance, and this is how some of them were first identified. When the ABC transport proteins are overexpressed in cancer cells, they can export anticancer drugs and render tumors resistant.[Scott MP, Lodish HF, Berk A, Kaiser, C, Krieger M, Bretscher A, Ploegh H, Amon A (2012). Molecular Cell Biology. San Francisco: W. H. Freeman. ISBN 978-1-4292-3413-9.]
This protein is a member of the MDR/TAP subfamily. Members of the MDR/TAP subfamily are involved in multidrug resistance as well as antigen presentation. This gene encodes a half-transporter involved in the transport of heme from the mitochondria to the cytosol. With iron/sulfur cluster precursors as its substrates, this protein may play a role in metal homeostasis.
Mutations in this gene have been implicated in X-linked sideroblastic anemia with ataxia.[“Entrez Gene: ABCB7 ATP-binding cassette, sub-family B (MDR/TAP), member 7”.]
ABCB7 has been shown to interact with Ferrochelatase.[Taketani S, Kakimoto K, Ueta H, Masaki R, Furukawa T (April 2003). “Involvement of ABC7 in the biosynthesis of heme in erythroid cells: interaction of ABC7 with ferrochelatase”. Blood. 101 (8): 3274–80. doi:10.1182/blood-2002-04-1212. PMID 12480705. S2CID 18599174.]
See also: ATP-binding cassette transporter
N-methylmesoporphyrin (N-MeMP) is a competitive inhibitor with protoporphyrin IX and is thought to be a transition state analog. As such, N-MeMP has been used extensively as a stabilizing ligand for x-ray crystallography structure determination.
- Medlock, A.; Swartz, L.; Dailey, T. A.; Dailey, H. A.; Lanzilotta, W. N. (29 January 2007). “Substrate interactions with human ferrochelatase”. Proceedings of the National Academy of Sciences. 104 (6): 1789–1793. Bibcode:2007PNAS..104.1789M. doi:10.1073/pnas.0606144104. PMC 1794275. PMID 17261801.
Frataxin acts as the Fe+2 chaperone and complexes with ferrochelatase on its mitochondrial matrix side.
- Bencze, Krisztina Z.; Yoon, Taejin; Mill?n-Pacheco, C?sar; Bradley, Patrick B.; Pastor, Nina; Cowan, J. A.; Stemmler, Timothy L. (2007). “Human frataxin: iron and ferrochelatase binding surface”. Chemical Communications (18): 1798–1800. doi:10.1039/B703195E. PMC 2862461. PMID 17476391.
Ferrochelatase can also insert other divalent metal ions into protoporphyrin. Some ions, such as Zn+2, Ni, and Co form other metalloporphyrins while heavier metal ions such as Mn, Pb, Hg, and Cd inhibit product release after metallation.
- Medlock, Amy E.; Carter, Michael; Dailey, Tamara A.; Dailey, Harry A.; Lanzilotta, William N. (October 2009). “Product Release Rather than Chelation Determines Metal Specificity for Ferrochelatase”. Journal of Molecular Biology. 393 (2): 308–319. doi:10.1016/j.jmb.2009.08.042. PMC 2771925. PMID 19703464.
See also
References
- “FECH – Ferrochelatase, mitochondrial precursor – Homo sapiens (Human) – FECH gene & protein”.
- Lecerof, D.; Fodje, M.; Hansson, A.; Hansson, M.; Al-Karadaghi, S. (March 2000). “Structural and mechanistic basis of porphyrin metallation by ferrochelatase”. Journal of Molecular Biology. 297 (1): 221–232. doi:10.1006/jmbi.2000.3569. PMID 10704318.
- Leeper, F. J. (1985). “The biosynthesis of porphyrins, chlorophylls, and vitamin B12”. Natural Product Reports. 2 (1): 19–47. doi:10.1039/NP9850200019. PMID 3895052.
- Berg, Jeremy; Tymoczko, John; Stryer, Lubert (2012). Biochemistry (7th ed.). New York: W.H. Freeman. ISBN 9781429229364.
- “RCSB PDB – 1Hrk: Crystal Structure of Human Ferrochelatase”.
- Wu, Chia-Kuei; Dailey, Harry A.; Rose, John P.; Burden, Amy; Sellers, Vera M.; Wang, Bi-Cheng (1 February 2001). “The 2.0 Å structure of human ferrochelatase, the terminal enzyme of heme biosynthesis”. Nature Structural Biology. 8 (2): 156–160. doi:10.1038/84152. PMID 11175906. S2CID 9822420.
- Karlberg, Tobias; Hansson, Mattias D.; Yengo, Raymond K.; Johansson, Renzo; Thorvaldsen, Hege O.; Ferreira, Gloria C.; Hansson, Mats; Al-Karadaghi, Salam (May 2008). “Porphyrin Binding and Distortion and Substrate Specificity in the Ferrochelatase Reaction: The Role of Active Site Residues”. Journal of Molecular Biology. 378 (5): 1074–1083. doi:10.1016/j.jmb.2008.03.040. PMC 2852141. PMID 18423489.
- Bencze, Krisztina Z.; Yoon, Taejin; Mill?n-Pacheco, C?sar; Bradley, Patrick B.; Pastor, Nina; Cowan, J. A.; Stemmler, Timothy L. (2007). “Human frataxin: iron and ferrochelatase binding surface”. Chemical Communications (18): 1798–1800. doi:10.1039/B703195E. PMC 2862461. PMID 17476391.
- James, William D.; Berger, Timothy G. (2006). Andrews’ Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- Rüfenacht, U.B.; Gouya, L.; Schneider-Yin, X.; Puy, H.; Schäfer, B.W.; Aquaron, R.; Nordmann, Y.; Minder, E.I.; Deybach, J.C. (1998). “Systematic Analysis of Molecular Defects in the Ferrochelatase Gene from Patients with Erythropoietic Protoporphyria”. The American Journal of Human Genetics. 62 (6): 1341–52. doi:10.1086/301870. PMC 1377149. PMID 9585598.
- “Lead Toxicity — What Are Possible Health Effects from Lead Exposure?”. Agency for Toxic Substances & Disease Registry. Retrieved 9 February 2021.
- Medlock, Amy E.; Shiferaw, Mesafint T.; Marcero, Jason R.; Vashisht, Ajay A.; Wohlschlegel, James A.; Phillips, John D.; Dailey, Harry A.; Liesa, Marc (19 August 2015). “Identification of the Mitochondrial Heme Metabolism Complex”. PLOS ONE. 10 (8): e0135896. Bibcode:2015PLoSO..1035896M. doi:10.1371/journal.pone.0135896. PMC 4545792. PMID 26287972.
- Chen, W.; Dailey, H. A.; Paw, B. H. (28 April 2010). “Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis”. Blood. 116 (4): 628–630. doi:10.1182/blood-2009-12-259614. PMC 3324294. PMID 20427704.
- Medlock, A.; Swartz, L.; Dailey, T. A.; Dailey, H. A.; Lanzilotta, W. N. (29 January 2007). “Substrate interactions with human ferrochelatase”. Proceedings of the National Academy of Sciences. 104 (6): 1789–1793. Bibcode:2007PNAS..104.1789M. doi:10.1073/pnas.0606144104. PMC 1794275. PMID 17261801.
- Medlock, Amy E.; Carter, Michael; Dailey, Tamara A.; Dailey, Harry A.; Lanzilotta, William N. (October 2009). “Product Release Rather than Chelation Determines Metal Specificity for Ferrochelatase”. Journal of Molecular Biology. 393 (2): 308–319. doi:10.1016/j.jmb.2009.08.042. PMC 2771925. PMID 19703464.
Further reading
- Cox TM (June 1997). “Erythropoietic protoporphyria”. Journal of Inherited Metabolic Disease. 20 (2): 258–69. doi:10.1023/A:1005317124985. PMID 9211198. S2CID 12493042.
- Brenner DA, Didier JM, Frasier F, Christensen SR, Evans GA, Dailey HA (June 1992). “A molecular defect in human protoporphyria”. American Journal of Human Genetics. 50 (6): 1203–10. PMC 1682545. PMID 1376018.
- Nakahashi Y, Fujita H, Taketani S, Ishida N, Kappas A, Sassa S (January 1992). “The molecular defect of ferrochelatase in a patient with erythropoietic protoporphyria”. Proceedings of the National Academy of Sciences of the United States of America. 89 (1): 281–5. Bibcode:1992PNAS…89..281N. doi:10.1073/pnas.89.1.281. PMC 48220. PMID 1729699.
- Lamoril J, Boulechfar S, de Verneuil H, Grandchamp B, Nordmann Y, Deybach JC (December 1991). “Human erythropoietic protoporphyria: two point mutations in the ferrochelatase gene”. Biochemical and Biophysical Research Communications. 181 (2): 594–9. doi:10.1016/0006-291X(91)91231-Z. PMID 1755842.
- Nakahashi Y, Taketani S, Okuda M, Inoue K, Tokunaga R (December 1990). “Molecular cloning and sequence analysis of cDNA encoding human ferrochelatase”. Biochemical and Biophysical Research Communications. 173 (2): 748–55. doi:10.1016/S0006-291X(05)80099-3. PMID 2260980.
- Rossi E, Attwood PV, Garcia-Webb P, Costin KA (May 1990). “Inhibition of human lymphocyte ferrochelatase activity by hemin”. Biochimica et Biophysica Acta (BBA) – Protein Structure and Molecular Enzymology. 1038 (3): 375–81. doi:10.1016/0167-4838(90)90251-A. PMID 2340297.
- Polson RJ, Lim CK, Rolles K, Calne RY, Williams R (September 1988). “The effect of liver transplantation in a 13-year-old boy with erythropoietic protoporphyria”. Transplantation. 46 (3): 386–9. doi:10.1097/00007890-198809000-00010. PMID 3047929.
- Bonkovsky HL, Schned AR (January 1986). “Fatal liver failure in protoporphyria. Synergism between ethanol excess and the genetic defect”. Gastroenterology. 90 (1): 191–201. doi:10.1016/0016-5085(86)90093-4. PMID 3940245.
- Prasad AR, Dailey HA (August 1995). “Effect of cellular location on the function of ferrochelatase”. The Journal of Biological Chemistry. 270 (31): 18198–200. doi:10.1074/jbc.270.31.18198. PMID 7629135.
- Sarkany RP, Alexander GJ, Cox TM (June 1994). “Recessive inheritance of erythropoietic protoporphyria with liver failure”. Lancet. 343 (8910): 1394–6. doi:10.1016/S0140-6736(94)92525-9. PMID 7910885. S2CID 42243172.
- Tugores A, Magness ST, Brenner DA (December 1994). “A single promoter directs both housekeeping and erythroid preferential expression of the human ferrochelatase gene”. The Journal of Biological Chemistry. 269 (49): 30789–97. doi:10.1016/S0021-9258(18)47351-6. PMID 7983009.
- Dailey HA, Sellers VM, Dailey TA (January 1994). “Mammalian ferrochelatase. Expression and characterization of normal and two human protoporphyric ferrochelatases”. The Journal of Biological Chemistry. 269 (1): 390–5. doi:10.1016/S0021-9258(17)42362-3. PMID 8276824.
- Wang X, Poh-Fitzpatrick M, Carriero D, Ostasiewicz L, Chen T, Taketani S, Piomelli S (April 1993). “A novel mutation in erythropoietic protoporphyria: an aberrant ferrochelatase mRNA caused by exon skipping during RNA splicing”. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. 1181 (2): 198–200. doi:10.1016/0925-4439(93)90112-e. PMID 8481408.
- Nakahashi Y, Miyazaki H, Kadota Y, Naitoh Y, Inoue K, Yamamoto M, Hayashi N, Taketani S (May 1993). “Molecular defect in human erythropoietic protoporphyria with fatal liver failure”. Human Genetics. 91 (4): 303–6. doi:10.1007/BF00217346. PMID 8500787. S2CID 5844599.
- Imoto S, Tanizawa Y, Sato Y, Kaku K, Oka Y (July 1996). “A novel mutation in the ferrochelatase gene associated with erythropoietic protoporphyria”. British Journal of Haematology. 94 (1): 191–7. doi:10.1046/j.1365-2141.1996.d01-1771.x. PMID 8757534. S2CID 27290533.
- Crouse BR, Sellers VM, Finnegan MG, Dailey HA, Johnson MK (December 1996). “Site-directed mutagenesis and spectroscopic characterization of human ferrochelatase: identification of residues coordinating the [2Fe-2S] cluster”. Biochemistry. 35 (50): 16222–9. doi:10.1021/bi9620114. PMID 8973195.
External links
- UMich Orientation of Proteins in Membranes protein/pdbid-1hrk
- Ferrochelatase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
| Enzymes involved in the metabolism of heme and porphyrin |
|---|
Leave a Reply