Agrin is a large chimeric proteoglycan, a heparan sulfate and chondroitin proteoglycan, whose best-characterised role is in the development of the neuromuscular junction during embryogenesis
Agrin was originally found in the electric organ of Tarpedo california and in the basal lamina at the neuromuscular junction as a protein that directs the aggregation of acetylcholine receptors (AChR) and acetylcholinesterase (AChE) at synaptic sites.
- Agrin at ScienceDirect https://www.sciencedirect.com/topics/medicine-and-dentistry/agrin

Tetronarce californica also known as the Pacific electric ray is a species of electric ray in the family Torpedinidae, endemic to the coastal waters of the northeastern Pacific Ocean from Baja California to British Columbia. It generally inhabits sandy flats, rocky reefs, and kelp forests from the surface to a depth of 200 m (660 ft), but has also been known to make forays into the open ocean. Measuring up to 1.4 m (4.6 ft) long, this species has smooth-rimmed spiracles (paired respiratory openings behind the eyes) and a dark gray, slate, or brown dorsal coloration, sometimes with dark spots. Its body form is typical of the genus, with a rounded pectoral fin disc wider than long and a thick tail bearing two dorsal fins of unequal size and a well-developed caudal fin. Solitary and nocturnal, the Pacific electric ray can generate up to 45 volts of electricity for the purposes of subduing prey or self-defense. It feeds mainly on bony fishes, ambushing them from the substrate during the day and actively hunting for them at night. Reproduction is aplacental viviparous, meaning that the embryos are initially nourished by yolk, later supplemented by histotroph (“uterine milk”) produced by the mother. Care should be exercised around the Pacific electric ray, as it has been known to act aggressively if provoked and its electric shock can potentially incapacitate a diver. It and other electric rays are used as model organisms for biomedical research because their electric organs contain an abundance of important nervous system proteins such as nicotinic acetylcholine receptor and acetylcholinesterase. In the 1970s and 1980s, acetylcholine receptors from this species and the marbled electric ray (T. marmorata) became the first neurotransmitter receptors to be isolated and sequenced, in what is considered to be a landmark success in the field of neurobiology. This led to a number of further advances, one of the most significant being the elucidation of the pathophysiology underlying the disease myasthenia gravis.
- Scandalios, J.G. & Wright, T.R.F. (1991). Advances in Genetics. Academic Press. p. 178. ISBN 0-12-017629-7.
- Bullock, T.H. (2005). Electroreception. Birkhäuser. p. 41. ISBN 0-387-23192-7.
- Histotrophy is a form of matrotrophy exhibited by some live-bearing sharks and rays, in which the developing embryo receives additional nutrition from its mother in the form of uterine secretions, known as histotroph (or “uterine milk”). It is one of the three major modes of elasmobranch reproduction encompassed by “aplacental viviparity“, and can be contrasted with yolk-sac viviparity (in which the embryo is solely sustained by yolk) and oophagy (in which the embryo feeds on ova). There are two categories of histotrophy:
- In mucoid or limited histotrophy, the developing embryo ingests uterine mucus or histotroph as a supplement to the energy supplies provided by its yolk sac. This form of histotrophy is known to occur in the dogfish sharks (Squaliformes) and the electric rays (Torpediniformes), and may be more widespread.
- In lipid histotrophy, the developing embryo is supplied with protein and lipid-enriched histotroph through specialized finger-like structures known as trophonemata. The additional nutrition provided by the enriched histotroph allows the embryo to increase in mass from the egg by several orders of magnitude by the time it is born, much greater than is possible in mucoid histotrophy. This form of histotrophy is found in stingrays and their relatives (Myliobatiformes).
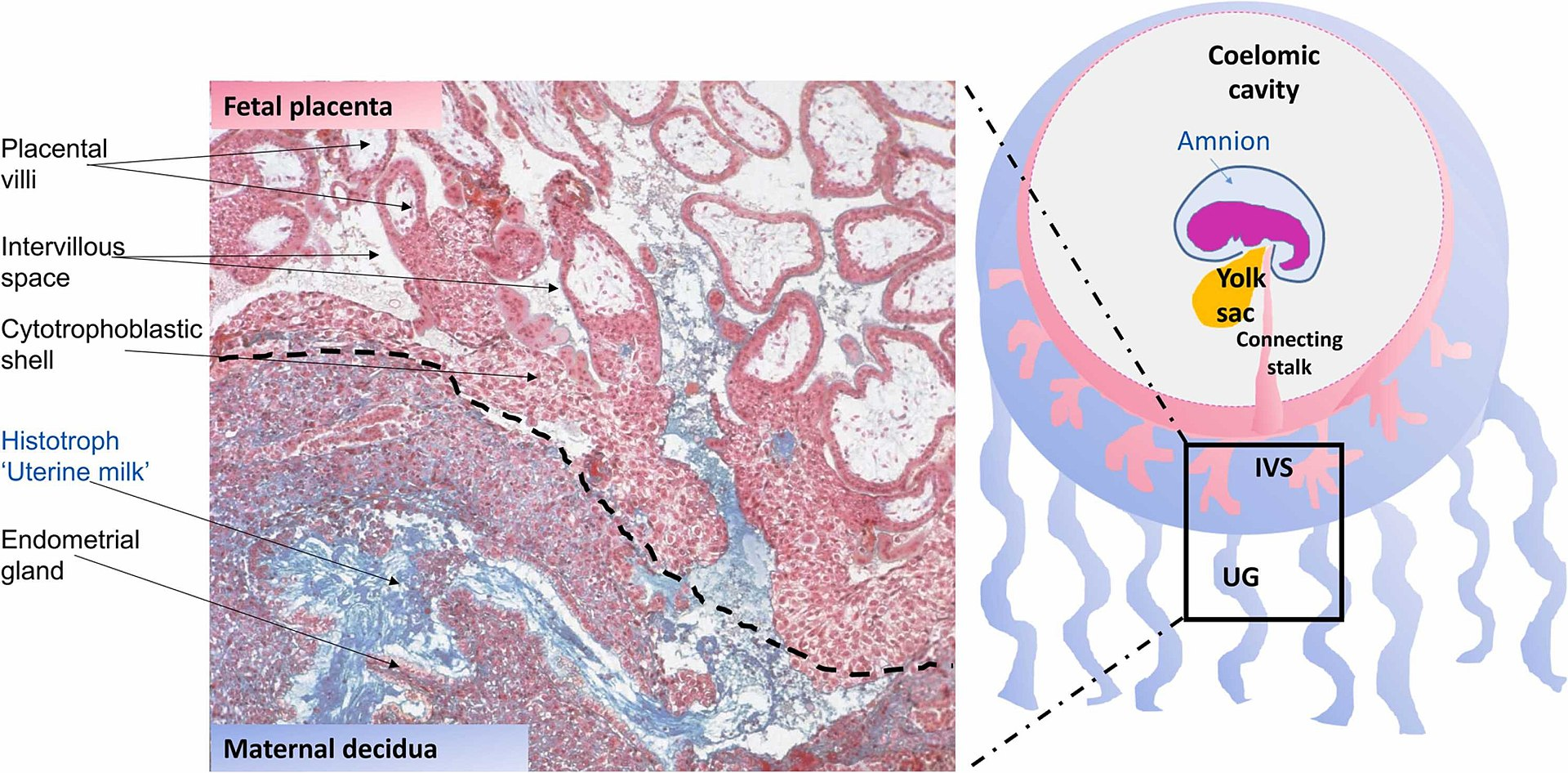
Uterine glands or endometrial glands are tubular glands, lined by a simple columnar epithelium, found in the functional layer of the endometrium that lines the uterus. Their appearance varies during the menstrual cycle. During the proliferative phase, uterine glands appear long due to estrogen secretion by the ovaries. During the secretory phase, the uterine glands become very coiled with wide lumens and produce a glycogen-rich secretion known as histotroph or uterine milk. This change corresponds with an increase in blood flow to spiral arteries due to increased progesterone secretion from the corpus luteum. During the pre-menstrual phase, progesterone secretion decreases as the corpus luteum degenerates, which results in decreased blood flow to the spiral arteries. The functional layer of the uterus containing the glands becomes necrotic, and eventually sloughs off during the menstrual phase of the cycle. They are of small size in the unimpregnated uterus, but shortly after impregnation become enlarged and elongated, presenting a contorted or waved appearance. Hormones produced in early pregnancy stimulate the uterine glands to secrete a number of substances to give nutrition and protection to the embryo and fetus, and the fetal membranes. These secretions are known as histiotroph, alternatively histotroph, and also as uterine milk. Important uterine milk proteins are glycodelin-A, and osteopontin. Some secretory components from the uterine glands are taken up by the secondary yolk sac lining the exocoelomic cavity during pregnancy, and may thereby assist in providing fetal nutrition.
- This article incorporates text in the public domain from page 1262 of the 20th edition of Gray’s Anatomy (1918)
- Burton, GJ; Cindrova-Davies, T; Turco, MY (December 2020). “Review: Histotrophic nutrition and the placental-endometrial dialogue during human early pregnancy”. Placenta. 102: 21–26. doi:10.1016/j.placenta.2020.02.008. PMID 33218574. S2CID 214025290.
- Kelleher, AM; DeMayo, FJ; Spencer, TE (1 October 2019). “Uterine Glands: Developmental Biology and Functional Roles in Pregnancy”. Endocrine Reviews. 40 (5): 1424–1445. doi:10.1210/er.2018-00281. PMC 6749889. PMID 31074826.
- Burton GJ, Watson AL, Hempstock J, Skepper JN, Jauniaux E (June 2002). “Uterine glands provide histiotrophic nutrition for the human fetus during the first trimester of pregnancy”. J. Clin. Endocrinol. Metab. 87 (6): 2954–9. doi:10.1210/jcem.87.6.8563. PMID 12050279.
- Swiss embryology (from UL, UB, and UF) gnidation/role02
- Histology at KUMC epithel-epith06
- Anatomy photo: Reproductive/mammal/uterus1/uterus2 – Comparative Organology at University of California, Davis – “Mammal, uterus (LM, Medium)”
- UIUC Histology Subject 1024
- Cole, K.S. (2010). Reproduction and Sexuality in Marine Fishes: Patterns and Processes. University of California Press. pp. 8–9. ISBN 978-0-520-26433-5.
- Hamlett, W.C., ed. (2005). Reproductive Biology and Phylogeny of Chondrichthyes: Sharks, Batoids and Chimaeras. Science Publishers. pp. 46–47. ISBN 1-57808-314-1.
The aggregating factor was named ‘agrin’ from the Greek word ‘ageirein’, which means ‘to assemble.’ In humans, this protein is encoded by the AGRN gene. Agrin is named based on its involvement in the aggregation of acetylcholine receptors during synaptogenesis.
- Rupp F, Payan DG, Magill-Solc C, Cowan DM, Scheller RH (May 1991). “Structure and expression of a rat agrin”. Neuron. 6 (5): 811–823. doi:10.1016/0896-6273(91)90177-2. PMID 1851019. S2CID 44440186.
- Kröger S, Schröder JE (October 2002). “Agrin in the developing CNS: new roles for a synapse organizer”. News in Physiological Sciences. 17 (5): 207–212. doi:10.1152/nips.01390.2002. PMID 12270958. S2CID 2988918.
- Groffen AJ, Buskens CA, van Kuppevelt TH, Veerkamp JH, Monnens LA, van den Heuvel LP (May 1998). “Primary structure and high expression of human agrin in basement membranes of adult lung and kidney”. European Journal of Biochemistry. 254 (1): 123–128. doi:10.1046/j.1432-1327.1998.2540123.x. PMID 9652404.
- Agrin at ScienceDirect https://www.sciencedirect.com/topics/medicine-and-dentistry/agrin
This protein has nine domains homologous to protease inhibitors. It may also have functions in other tissues and during other stages of development. It is a major proteoglycan component in the glomerular basement membrane and may play a role in the renal filtration and cell-matrix interactions.
- Tsen G, Halfter W, Kröger S, Cole GJ (February 1995). “Agrin is a heparan sulfate proteoglycan”. The Journal of Biological Chemistry. 270 (7): 3392–3399. doi:10.1074/jbc.270.7.3392. PMID 7852425.
- Groffen AJ, Ruegg MA, Dijkman H, van de Velden TJ, Buskens CA, van den Born J, et al. (January 1998). “Agrin is a major heparan sulfate proteoglycan in the human glomerular basement membrane”. The Journal of Histochemistry and Cytochemistry. 46 (1): 19–27. doi:10.1177/002215549804600104. PMID 9405491. S2CID 24353753.
Agrin functions by activating the MuSK protein (for Muscle-Specific Kinase), which is a receptor tyrosine kinase required for the formation and maintenance of the neuromuscular junction. Agrin is required to activate MuSK. Agrin is also required for neuromuscular junction formation.
- Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR, Thomas S, LeBeau MM, Fernald AA, Copeland NG, Jenkins NA, Burden SJ, Glass DJ, Yancopoulos GD (Sep 1995). “Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury”. Neuron. 15 (3): 573–584. doi:10.1016/0896-6273(95)90146-9. PMID 7546737.
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD (May 1996). “The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo”. Cell. 85 (4): 501–512. doi:10.1016/s0092-8674(00)81251-9. PMID 8653786.
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD (May 1996). “Agrin acts via a MuSK receptor complex”. Cell. 85 (4): 513–523. doi:10.1016/s0092-8674(00)81252-0. PMID 8653787.
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (May 1996). “Defective neuromuscular synaptogenesis in agrin-deficient mutant mice”. Cell. 85 (4): 525–535. doi:10.1016/s0092-8674(00)81253-2. PMID 8653788
MuSK (for Muscle-Specific Kinase) is a receptor tyrosine kinase required for the formation and maintenance of the neuromuscular junction. It is activated by a nerve-derived proteoglycan called agrin, which is similarly also required for neuromuscular junction formation.
- Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR, Thomas S, LeBeau MM, Fernald AA, Copeland NG, Jenkins NA, Burden SJ, Glass DJ, Yancopoulos GD (Sep 1995). “Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury”. Neuron. 15 (3): 573–584. doi:10.1016/0896-6273(95)90146-9. PMID 7546737. S2CID 17575761.
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD (May 1996). “The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo”. Cell. 85 (4): 501–512. doi:10.1016/s0092-8674(00)81251-9. PMID 8653786. S2CID 17455481.
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD (May 1996). “Agrin acts via a MuSK receptor complex”. Cell. 85 (4): 513–523. doi:10.1016/s0092-8674(00)81252-0. PMID 8653787. S2CID 14930468.
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (May 1996). “Defective neuromuscular synaptogenesis in agrin-deficient mutant mice”. Cell. 85 (4): 525–535. doi:10.1016/s0092-8674(00)81253-2. PMID 8653788. S2CID 12517490.
Upon activation by its ligand agrin, MuSK signals via the proteins called casein kinase 2 (CK2), Dok-7 and rapsyn, to induce “clustering” of acetylcholine receptors (AChR). Both CK2 and Dok-7 are required for MuSK-induced formation of the neuromuscular junction, since mice lacking Dok-7 failed to form AChR clusters or neuromuscular synapses, and since downregulation of CK2 also impedes recruitment of AChR to the primary MuSK scaffold. In addition to the proteins mentioned, other proteins are then gathered, to form the endplate to the neuromuscular junction. The nerve terminates onto the endplate, forming the neuromuscular junction – a structure required to transmit nerve impulses to the muscle, and thus initiating muscle contraction.
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD (May 1996). “Agrin acts via a MuSK receptor complex”. Cell. 85 (4): 513–523. doi:10.1016/s0092-8674(00)81252-0. PMID 8653787. S2CID 14930468.
- Cheusova T, Khan MA, Schubert SW, Gavin AC, Buchou T, Jacob G, Sticht H, Allende J, Boldyreff B, Brenner HR, Hashemolhosseini S (Jul 2006). “Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction”. Genes & Development. 20 (13): 1800–16. doi:10.1101/gad.375206. PMC 1522076. PMID 16818610.
- Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, Kubo S, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y (Jun 2006). “The muscle protein Dok-7 is essential for neuromuscular synaptogenesis”. Science. 312 (5781): 1802–5. Bibcode:2006Sci…312.1802O. doi:10.1126/science.1127142. PMID 16794080. S2CID 45730054.
Antibodies directed against this protein (Anti-MuSK autoantibodies) are found in some people with myasthenia gravis not demonstrating antibodies to the acetylcholine receptor. The disease still causes loss of acetylcholine receptor activity, but the symptoms affected people experience may differ from those of people with other causes of myasthenia gravis.[citation needed]
- Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A (Mar 2001). “Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies”. Nature Medicine. 7 (3): 365–8. doi:10.1038/85520. PMID 11231638. S2CID 18641849.
- Barrett-Jolley R, Byrne N, Vincent A, Newsom-Davis J (Oct 1994). “Plasma from patients with seronegative myasthenia gravis inhibit nAChR responses in the TE671/RD cell line”. Pflügers Archiv. 428 (5–6): 492–8. doi:10.1007/BF00374570. PMID 7838671. S2CID 5611563.
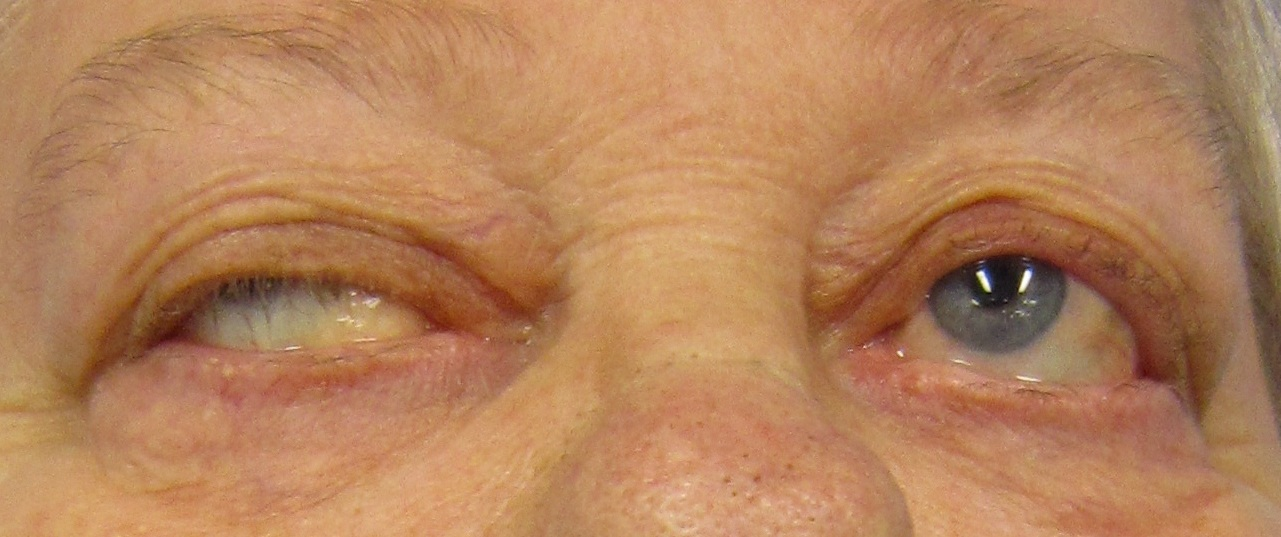
Myasthenia gravis (MG) is a long-term neuromuscular junction disease that leads to varying degrees of skeletal muscle weakness. The most commonly affected muscles are those of the eyes, face, and swallowing. It can result in double vision, drooping eyelids, and difficulties in talking and walking. Onset can be sudden. Those affected often have a large thymus or develop a thymoma. Myasthenia gravis is an autoimmune disease of the neuro-muscular junction which results from antibodies that block or destroy nicotinic acetylcholine receptors (AChR) at the junction between the nerve and muscle. This prevents nerve impulses from triggering muscle contractions. Most cases are due to immunoglobulin G1 (IgG1) and IgG3 antibodies that attack AChR in the postsynaptic membrane, causing complement-mediated damage and muscle weakness. Rarely, an inherited genetic defect in the neuromuscular junction results in a similar condition known as congenital myasthenia.
- Myasthenia Gravis Fact Sheet”. National Institute of Neurological Disorders and Stroke.
- Kahan S (2005). In a Page: Neurology. Lippincott Williams & Wilkins. p. 118. ISBN 978-1-4051-0432-6. Archived from the original on 8 September 2017.
- Kaminski HJ (2009). Myasthenia Gravis and Related Disorders (2 ed.). Springer Science & Business Media. p. 72. ISBN 978-1-59745-156-7. Archived from the original on 8 September 2017.
- Adams JG (2012). Emergency Medicine: Clinical Essentials (2 ed.). Elsevier Health Sciences. p. 844. ISBN 978-1-4557-3394-1. Archived from the original on 8 September 2017.
- Salari N, Fatahi B, Bartina Y, Kazeminia M, Fatahian R, Mohammadi P, et al. (December 2021). “Global prevalence of myasthenia gravis and the effectiveness of common drugs in its treatment: a systematic review and meta-analysis”. Journal of Translational Medicine. 19 (1): 516. doi:10.1186/s12967-021-03185-7. PMC 8686543. PMID 34930325.
- Young C, McGill SC (April 2021). Rituximab for the Treatment of Myasthenia Gravis: A 2021 Update [Internet] (Report). Ottawa (ON): Canadian Agency for Drugs and Technologies in Health. PMID 34255447.
- Dabi A, Solieman N, Kurukumbi M, Kalyanam J (2012). “Myasthenia Gravis: A Review”. Autoimmune Diseases. 2012: 1–10. doi:10.1155/2012/874680. PMC 3501798. PMID 23193443.
- Phillips WD, Vincent A (1 January 2016). “Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms”. F1000Research. 5: 1513. doi:10.12688/f1000research.8206.1. PMC 4926737. PMID 27408701.
- Kandel E, Schwartz J, Jessel T, Siegelbaum S, Hudspeth A (2012). Principles of Neural Science (5 ed.). pp. 318–319.
- Vrinten C, van der Zwaag AM, Weinreich SS, Scholten RJ, Verschuuren JJ (December 2014). “Ephedrine for myasthenia gravis, neonatal myasthenia and the congenital myasthenic syndromes”. The Cochrane Database of Systematic Reviews. 2014 (12): CD010028. doi:10.1002/14651858.CD010028.pub2. PMC 7387729. PMID 25515947.
| Class | Description |
|---|---|
| I | Any eye muscle weakness, possible ptosis, no other evidence of muscle weakness elsewhere |
| II | Eye muscle weakness of any severity, mild weakness of other muscles |
| IIa | Predominantly limb or axial muscles |
| IIb | Predominantly bulbar and/or respiratory muscles |
| III | Eye muscle weakness of any severity, moderate weakness of other muscles |
| IIIa | Predominantly limb or axial muscles |
| IIIb | Predominantly bulbar and/or respiratory muscles |
| IV | Eye muscle weakness of any severity, severe weakness of other muscles |
| IVa | Predominantly limb or axial muscles |
| IVb | Predominantly bulbar and/or respiratory muscles |
| V | Intubation needed to maintain airway |
Discovery
Agrin was first identified by the U.J. McMahan laboratory, Stanford University.
- Magill C, Reist NE, Fallon JR, Nitkin RM, Wallace BG, McMahan UJ (1987). “Chapter 32 Agrin”. Neural Regeneration. Progress in Brain Research. Vol. 71. pp. 391–396. doi:10.1016/S0079-6123(08)61840-3. ISBN 978-0-444-80814-1. PMID 3035610
Mechanism of action
During development in humans, the growing end of motor neuron axons secrete a protein called agrin. When secreted, agrin binds to several receptors on the surface of skeletal muscle. The receptor which appears to be required for the formation of the neuromuscular junction (NMJ) is called the MuSK receptor (Muscle specific kinase). MuSK is a receptor tyrosine kinase – meaning that it induces cellular signaling by causing the addition of phosphate molecules to particular tyrosines on itself and on proteins that bind the cytoplasmic domain of the receptor.
- Sanes JR, Lichtman JW (November 2001). “Induction, assembly, maturation and maintenance of a postsynaptic apparatus”. Nature Reviews. Neuroscience. 2 (11): 791–805. doi:10.1038/35097557. PMID 11715056. S2CID 52802445.
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, et al. (May 1996). “Agrin acts via a MuSK receptor complex”. Cell. 85 (4): 513–523. doi:10.1016/S0092-8674(00)81252-0. PMID 8653787. S2CID 14930468.
- Sanes JR, Apel ED, Gautam M, Glass D, Grady RM, Martin PT, et al. (May 1998). “Agrin receptors at the skeletal neuromuscular junction”. Annals of the New York Academy of Sciences. 841 (1): 1–13. Bibcode:1998NYASA.841….1S. doi:10.1111/j.1749-6632.1998.tb10905.x. PMID 9668217. S2CID 20097480
In addition to MuSK, agrin binds several other proteins on the surface of muscle, including dystroglycan and laminin. It is seen that these additional binding steps are required to stabilize the NMJ.
The requirement for Agrin and MuSK in the formation of the NMJ was demonstrated primarily by knockout mouse studies. In mice that are deficient for either protein, the neuromuscular junction does not form. Many other proteins also comprise the NMJ, and are required to maintain its integrity. For example, MuSK also binds a protein called “dishevelled” (Dvl), which is in the Wnt signalling pathway. Dvl is additionally required for MuSK-mediated clustering of AChRs, since inhibition of Dvl blocks clustering.
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (May 1996). “Defective neuromuscular synaptogenesis in agrin-deficient mutant mice”. Cell. 85 (4): 525–535. doi:10.1016/S0092-8674(00)81253-2. PMID 8653788. S2CID 12517490.
Dishevelled (Dsh) is a family of proteins involved in canonical and non-canonical Wnt signalling pathways. Dsh (Dvl in mammals) is a cytoplasmic phosphoprotein that acts directly downstream of frizzled receptors. It takes its name from its initial discovery in flies, where a mutation in the dishevelled gene was observed to cause improper orientation of body and wing hairs. There are vertebrate homologs in zebrafish, Xenopus (Xdsh), mice (Dvl1, -2, -3) and humans (DVL-1, -2, -3). Dsh relays complex Wnt signals in tissues and cells, in normal and abnormal contexts. It is thought to interact with the SPATS1 protein when regulating the Wnt Signalling pathway.
- Penton A, Wodarz A, Nusse R (June 2002). “A mutational analysis of dishevelled in Drosophila defines novel domains in the dishevelled protein as well as novel suppressing alleles of axin”. Genetics. 161 (2): 747–62. doi:10.1093/genetics/161.2.747. PMC 1462152. PMID 12072470.
- Wallingford JB, Habas R (October 2005). “The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity”. Development. 132 (20): 4421–36. doi:10.1242/dev.02068. PMID 16192308.
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363
- Zhang H, Zhang H, Zhang Y, Ng SS, Ren F, Wang Y, Duan Y, Chen L, Zhai Y, Guo Q, Chang Z (November 2010). “Dishevelled-DEP domain interacting protein (DDIP) inhibits Wnt signaling by promoting TCF4 degradation and disrupting the TCF4/beta-catenin complex”. Cellular Signalling. 22 (11): 1753–60. doi:10.1016/j.cellsig.2010.06.016. PMID 20603214.
Spermatogenesis associated serine rich 1 (SPATS1) is a protein which in humans is encoded by the SPATS1 gene. It is also known by the aliases Dishevelled-DEP domain interacting protein (DDIP), Spermatogenesis Associated 8 (SPATA8), and serin-rich spermatogenic protein 1 (SRSP1). A general idea of its chemical structure, subcellular localization, expression, and conservation is known. Research suggests SPATS1 may play a role in the canonical Wnt Signaling pathway and in the first spermatogenic wave. The expression of this protein has been found to greatly decline in adulthood, compared to expression levels measured in fetuses. Studies have shown some fluctuation during the gestation period, but overall remaining relatively high. There has also been evidence of high expression levels up until day 28 postpartum. Expression of this protein has been found in peritubular myoid cells, gonocytes, pachytene spermatocytes, spermatogonia, myoid cells, and Sertoli cells.
- Capoano CA, Wettstein R, Kun A, Geisinger A (2010). “Spats 1 (Srsp1) is differentially expressed during testis development of the rat”. Gene Expression Patterns. 10 (1): 1–8. doi:10.1016/j.gep.2009.11.006. PMID 19948251.
- “GEO Profiles”.
- “Homo sapiens spermatogenesis associated serine rich 1 (SPATS1), mRNA – Nucleotide – NCBI”. www.ncbi.nlm.nih.gov. Retrieved 2017-02-20.

Mouse brains have shown expression in various areas of the brain including the pituitary gland, the prefrontal cortex, the frontal lobe, the cerebellum, and the parietal lobe. Highest expression levels have been found in the testes, the next highest levels being found in the trachea. A protein abundance histogram, which compares the abundance of a desired protein to other proteins, shows that SPATS1 is on the lower level of expression.
- “Allen Brain”.
- “Homo sapiens spermatogenesis associated serine rich 1 (SPATS1), mRNA – Nucleotide – NCBI”. www.ncbi.nlm.nih.gov. Retrieved 2017-02-20.
Possible interacting proteins are listed in the table below. Note that these proteins have not been experimentally confirmed to interact with SPATS1. Instead, their interaction potential was determined by looking at concurrence patterns and textmining.
| Protein Name | Function | Score |
|---|---|---|
| zinc finger protein 683 aka ZNF683 | may be involved in transcriptional regulation | 0.633 |
| transmembrane channel like 5 aka TMC5 | probable ion channel | 0.624 |
| gametocyte specific factor 1 like aka GTSF1L | unknown | 0.567 |
| transmembrane protein 225 aka TMEM225 | most likely inhibits phosphate 1 (PP1) in sperm via binding to catalytic sub-unit PPP1CC | 0.566 |
| spermatogenesis associated 3 aka SPATA3 | unknown | 0.537 |
| family with sequence similarity71 member F1 aka FAM71F1 | unknown | 0.535 |
| chromosome 9 open reading frame 139 aka C9orf139 | unknown | 0.477 |
| sperm acrosome associated 4 aka SPACA4 | sperm surface membrane protein that may be involved in sperm – egg plasma membrane adhesion and fusion during fertilization | 0.472 |
| sex comb on midleg-like protein 4 aka SCML4 | PcG proteins that act by forming multi-protein complexes, which are required to maintain the transcriptionally repressive state of homeotic genes throughout development | 0.457 |
Dishevelled plays important roles in both the embryo and the adult, ranging from cellular differentiation and cell polarity to social behavior.
- Wallingford JB, Habas R (October 2005). “The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity”. Development. 132 (20): 4421–36. doi:10.1242/dev.02068. PMID 16192308.
Members
There are three human genes that encode for the dishevelled proteins:
- DVL1
- Segment polarity protein dishevelled homolog DVL-1 is a protein that in humans is encoded by the DVL1 gene.
- Pizzuti A, Amati F, Calabrese G, Mari A, Colosimo A, Silani V, Giardino L, Ratti A, Penso D, Calzà L, Palka G, Scarlato G, Novelli G, Dallapiccola B (Jan 1997). “cDNA characterization and chromosomal mapping of two human homologues of the Drosophila dishevelled polarity gene”. Hum Mol Genet. 5 (7): 953–8. doi:10.1093/hmg/5.7.953. PMID 8817329.
- “Entrez Gene: DVL1 dishevelled, dsh homolog 1 (Drosophila)”.
- DVL1, the human homolog of the Drosophila dishevelled gene (dsh) encodes a cytoplasmic phosphoprotein that regulates cell proliferation, acting as a transducer molecule for developmental processes, including segmentation and neuroblast specification. DVL1 is a candidate gene for processes involved in cell transformations involved in neuroblastoma. The Schwartz-Jampel syndrome and Charcot-Marie-Tooth disease type 2A have been mapped to the same region as DVL1. The phenotypes of these diseases may be consistent with defects which might be expected from aberrant expression of a DVL gene during development. Three transcript variants encoding three different isoforms have been found for this gene.
- DVL1 has been shown to interact with: AXIN1, DVL3, EPS8, and Mothers against decapentaplegic homolog 3.
- Li L, Yuan H, Weaver CD, Mao J, Farr GH, Sussman DJ, Jonkers J, Kimelman D, Wu D (Aug 1999). “Axin and Frat1 interact with dvl and GSK, bridging Dvl to GSK in Wnt-mediated regulation of LEF-1”. EMBO J. 18 (15): 4233–40. doi:10.1093/emboj/18.15.4233. PMC 1171499. PMID 10428961.
- Kim MJ, Chia IV, Costantini F (Nov 2008). “SUMOylation target sites at the C terminus protect Axin from ubiquitination and confer protein stability”. FASEB J. 22 (11): 3785–94. doi:10.1096/fj.08-113910. PMC 2574027. PMID 18632848.
- Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A (Jun 1999). “DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability”. Mol. Cell. Biol. 19 (6): 4414–22. doi:10.1128/mcb.19.6.4414. PMC 104400. PMID 10330181.
- Inobe M, Katsube Ki, Miyagoe Y, Nabeshima Yi, Takeda S (Dec 1999). “Identification of EPS8 as a Dvl1-associated molecule”. Biochem. Biophys. Res. Commun. 266 (1): 216–21. doi:10.1006/bbrc.1999.1782. PMID 10581192.
- Warner DR, Pisano MM, Roberts EA, Greene RM (Mar 2003). “Identification of three novel Smad binding proteins involved in cell polarity”. FEBS Lett. 539 (1–3): 167–73. doi:10.1016/s0014-5793(03)00155-8. PMID 12650946.
- Segment polarity protein dishevelled homolog DVL-1 is a protein that in humans is encoded by the DVL1 gene.
- DVL2
- Segment polarity protein dishevelled homolog DVL-2 is a protein that in humans is encoded by the DVL2 gene.
- Greco TL, Sussman DJ, Camper SA (January 1997). “Dishevelled-2 maps to human chromosome 17 and distal to Wnt3a and vestigial tail (vt) on mouse chromosome 11”. Mamm Genome. 7 (6): 475–6. doi:10.1007/s003359900144. PMID 8662242. S2CID 39619679.
- “Entrez Gene: DVL2 dishevelled, dsh homolog 2 (Drosophila)”
- This gene encodes a member of the dishevelled (dsh) protein family. The vertebrate dsh proteins have approximately 40% amino acid sequence similarity with Drosophila dsh. This gene encodes a 90-kD protein that undergoes posttranslational phosphorylation to form a 95-kD cytoplasmic protein, which may play a role in the signal transduction pathway mediated by multiple Wnt proteins. The mechanisms of dishevelled function in Wnt signaling are likely to be conserved among metazoans.
- DVL2 has been shown to interact with Zinc finger protein 165, DAB2 and Arrestin beta 1.
- Rual, Jean-François; Venkatesan Kavitha; Hao Tong; Hirozane-Kishikawa Tomoko; Dricot Amélie; Li Ning; Berriz Gabriel F; Gibbons Francis D; Dreze Matija; Ayivi-Guedehoussou Nono; Klitgord Niels; Simon Christophe; Boxem Mike; Milstein Stuart; Rosenberg Jennifer; Goldberg Debra S; Zhang Lan V; Wong Sharyl L; Franklin Giovanni; Li Siming; Albala Joanna S; Lim Janghoo; Fraughton Carlene; Llamosas Estelle; Cevik Sebiha; Bex Camille; Lamesch Philippe; Sikorski Robert S; Vandenhaute Jean; Zoghbi Huda Y; Smolyar Alex; Bosak Stephanie; Sequerra Reynaldo; Doucette-Stamm Lynn; Cusick Michael E; Hill David E; Roth Frederick P; Vidal Marc (October 2005). “Towards a proteome-scale map of the human protein-protein interaction network”. Nature. 437 (7062): 1173–8. Bibcode:2005Natur.437.1173R. doi:10.1038/nature04209. PMID 16189514. S2CID 4427026.
- Hocevar, B A; Mou F; Rennolds J L; Morris S M; Cooper J A; Howe P H (June 2003). “Regulation of the Wnt signaling pathway by disabled-2 (Dab2)”. EMBO J. 22 (12): 3084–94. doi:10.1093/emboj/cdg286. ISSN 0261-4189. PMC 162138. PMID 12805222.
- Chen, W; Hu L A; Semenov M V; Yanagawa S; Kikuchi A; Lefkowitz R J; Miller W E (December 2001). “β-Arrestin1 modulates lymphoid enhancer factor transcriptional activity through interaction with phosphorylated dishevelled proteins”. Proc. Natl. Acad. Sci. U.S.A. 98 (26): 14889–94. Bibcode:2001PNAS…9814889C. doi:10.1073/pnas.211572798. ISSN 0027-8424. PMC 64954. PMID 11742073.
- Segment polarity protein dishevelled homolog DVL-2 is a protein that in humans is encoded by the DVL2 gene.
- DVL3
- Segment polarity protein dishevelled homolog DVL-3 is a protein that in humans is encoded by the DVL3 gene.
- Pizzuti A, Amati F, Calabrese G, Mari A, Colosimo A, Silani V, Giardino L, Ratti A, Penso D, Calza L, Palka G, Scarlato G, Novelli G, Dallapiccola B (January 1997). “cDNA characterization and chromosomal mapping of two human homologues of the Drosophila dishevelled polarity gene”. Hum Mol Genet. 5 (7): 953–958. doi:10.1093/hmg/5.7.953. PMID 8817329.
- “Entrez Gene: DVL3 dishevelled, dsh homolog 3 (Drosophila)”
- This gene is a member of a multi-gene family which shares strong similarity with the Drosophila dishevelled gene, dsh. The Drosophila dishevelled gene encodes a cytoplasmic phosphoprotein that regulates cell proliferation.
- DVL3 has been shown to interact with DAB2, DVL1 and PRPF3.
- Hocevar, B A; Mou F; Rennolds J L; Morris S M; Cooper J A; Howe P H (June 2003). “Regulation of the Wnt signaling pathway by disabled-2 (Dab2)”. EMBO J. England. 22 (12): 3084–3094. doi:10.1093/emboj/cdg286. ISSN 0261-4189. PMC 162138. PMID 12805222.
- Kishida, S; Yamamoto H; Hino S; Ikeda S; Kishida M; Kikuchi A (June 1999). “DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta-catenin stability”. Mol. Cell. Biol. UNITED STATES. 19 (6): 4414–4422. doi:10.1128/mcb.19.6.4414. ISSN 0270-7306. PMC 104400. PMID 10330181.
- Rual, Jean-François; Venkatesan Kavitha; Hao Tong; Hirozane-Kishikawa Tomoko; Dricot Amélie; Li Ning; Berriz Gabriel F; Gibbons Francis D; Dreze Matija; Ayivi-Guedehoussou Nono; Klitgord Niels; Simon Christophe; Boxem Mike; Milstein Stuart; Rosenberg Jennifer; Goldberg Debra S; Zhang Lan V; Wong Sharyl L; Franklin Giovanni; Li Siming; Albala Joanna S; Lim Janghoo; Fraughton Carlene; Llamosas Estelle; Cevik Sebiha; Bex Camille; Lamesch Philippe; Sikorski Robert S; Vandenhaute Jean; Zoghbi Huda Y; Smolyar Alex; Bosak Stephanie; Sequerra Reynaldo; Doucette-Stamm Lynn; Cusick Michael E; Hill David E; Roth Frederick P; Vidal Marc (October 2005). “Towards a proteome-scale map of the human protein-protein interaction network”. Nature. England. 437 (7062): 1173–1178. Bibcode:2005Natur.437.1173R. doi:10.1038/nature04209. PMID 16189514. S2CID 4427026.
- Segment polarity protein dishevelled homolog DVL-3 is a protein that in humans is encoded by the DVL3 gene.
- Lee YN, Gao Y, Wang HY (February 2008). “Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, -2, and -3”. Cellular Signalling. 20 (2): 443–52. doi:1
Function
DVL is an integral part of the Wnt canonical pathway (β-catenin dependent) and non-canonical pathway (β-catenin-independent). In either of these, DVL acts downstream of a Frizzled receptor, although the pathways are distinct.
- Wallingford JB, Habas R (October 2005). “The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity”. Development. 132 (20): 4421–36. doi:10.1242/dev.02068. PMID 16192308.
- Mlodzik M (2016). The Dishevelled Protein Family: Still Rather a Mystery After Over 20 Years of Molecular Studies. Vol. 117. pp. 75–91. doi:10.1016/bs.ctdb.2015.11.027. ISBN 9780128013823. PMC 4939608. PMID 26969973
Wnt canonical pathway
The Wnt canonical pathway, also known as the Wnt/β-catenin pathway, is activated during development, regulation, cell differentiation, and proliferation. The Wnt canonical pathway moves DVL between the cytoplasm and nucleus, via a conserved nuclear export sequence (NES) and a nuclear localization sequence (NLS), both necessary for proper functioning. The binding of Wnt to Frizzled receptors helps recruit DVL to the membrane, providing a site for Axin and GSK3β to bind and phosphorylate LRP5/6 (transmembrane low-density lipoprotein receptor-related protein), preventing constitutive degradation of β-catenin. The prevention of this degradation DVL allows for β-catenin buildup in the nucleus, where it acts as a coactivator for TCF (T cell factor) to activate Wnt responsive genes. Conversely, without Wnt signaling, the destruction complex, made of APC, CKI, GSK3β and Axin, degrades β-catenin buildup, keeping the concentration of β-catenin in the cell low.
- Gómez-Orte E, Sáenz-Narciso B, Moreno S, Cabello J (September 2013). “Multiple functions of the noncanonical Wnt pathway”. Trends in Genetics. 29 (9): 545–53. doi:10.1016/j.tig.2013.06.003. PMID 23846023
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363
- Mlodzik M (2016). The Dishevelled Protein Family: Still Rather a Mystery After Over 20 Years of Molecular Studies. Vol. 117. pp. 75–91. doi:10.1016/bs.ctdb.2015.11.027. ISBN 9780128013823. PMC 4939608. PMID 26969973
Wnt non-canonical pathways
Planar cell polarity pathway
The planar cell polarity pathway (PCP) is the most notable β-catenin independent pathway – the Wnt signal is received by the Frizzled receptor, which relays signals to DVL, which then acts as a branch point for two independent pathways, leading to the activation of small GTPases Rho and Rac. For the Rho branch, Wnt signals induce DVL to form a complex with Daam1 (Dishevelled associated activator of morphogenesis 1). This complex then interacts with Rho guanine nucleotide exchange factor WGEF (weak-similarity GEF), which activates downstream effectors like Rho GTPase and Rho-associated kinase (ROCK), which activates actin and cytoskeleton architecture in the cell. For the Rac branch, DVL activates the Rac GTPase. Activating the Rac GTPase stimulates the downstream effector c-Jun N-terminal kinase (JNK), which controls rearrangements in the cytoskeleton and gene expression. More specifically, it regulates the polarity and movement of a cell, in processes in vertebrates (like Xenopus) including gastrulation, neural tube closure, and stereocilia orientation in the inner ear.
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363
- Gómez-Orte E, Sáenz-Narciso B, Moreno S, Cabello J (September 2013). “Multiple functions of the noncanonical Wnt pathway”. Trends in Genetics. 29 (9): 545–53. doi:10.1016/j.tig.2013.06.003. PMID 23846023
Wnt-calcium pathway
Another pathway independent of β-catenin is the Wnt-Ca2+ pathway, which is involved in cancer, inflammation, and neurodegeneration. Wnt triggers Frizzled-mediated activation, triggering a cascade leading to Ca2+ release, which activates effectors (e.g. CaMKII) that control gene transcription relevant to cell fate and cell migration. This pathway can switch off the Wnt/β-catenin cascade and it can also be inhibited by DVL activation.
- Gómez-Orte E, Sáenz-Narciso B, Moreno S, Cabello J (September 2013). “Multiple functions of the noncanonical Wnt pathway”. Trends in Genetics. 29 (9): 545–53. doi:10.1016/j.tig.2013.06.003. PMID 23846023.
- Gao C, Chen YG (May 2010). “Dishevelled: The hub of Wnt signaling”. Cellular Signalling. 22 (5): 717–27. doi:10.1016/j.cellsig.2009.11.021. PMID 20006983.
There are five main highly conserved regions that exist in all variations of DVL. These include an amino-terminal DIX (N-terminus) domain, a PDZ (central) domain, a carboxyl-terminal DEP (C-terminus) domain, and two regions with positively charged amino acid residues. There is a proline-heavy region between the DIX and PDZ domains, and a largely basic region between the DIX and PDZ domains that has conserved serine and threonine residues. These regions mediate protein-protein interactions and help DVL channel signals into either the β-catenin or the β-catenin independent pathways. Additionally, there is the conserved nuclear export sequence (NES) and a nuclear localization sequence (NLS), whose ability to move DVL between the cytoplasm and the nucleus may be an important part of its function.
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363

DIX domain (Dishevelled-Axin)
Located near the N-terminus region of DVL and consisting of about 82-85 amino acids for human DVL protein, DIX is found in proteins like Axin and coiled-coil protein DIX-domain-containing I (DIXdc1 or Ccd1). The DIX domain of DVL has five β-strands and one α helix with highly conserved amino acid residues.
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363
- Mlodzik M (2016). The Dishevelled Protein Family: Still Rather a Mystery After Over 20 Years of Molecular Studies. Vol. 117. pp. 75–91. doi:10.1016/bs.ctdb.2015.11.027. ISBN 9780128013823. PMC 4939608. PMID 26969973
PDZ domain
PDZ, whose name consists of the initials of first three identified proteins to share this common structural domain (Post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and Zonula occludens-1 protein (zo-1)), lies in the central region of DVL. PDZ typically has about 73 amino acids in each human DVL protein, and consists of 5-6 β-strands and 2-3 α-helices This motif plays a critical role in ligand binding and conformational properties of the DVL protein. This region mediates many protein-protein interactions and regulates multiple biological processes.
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363
- Mlodzik M (2016). The Dishevelled Protein Family: Still Rather a Mystery After Over 20 Years of Molecular Studies. Vol. 117. pp. 75–91. doi:10.1016/bs.ctdb.2015.11.027. ISBN 9780128013823. PMC 4939608. PMID 26969973
DEP domain (Dishevelled-EGL-10-Pleackstrin)
DEP, which is in C-terminal domain of DVL, has 75 amino acids in the human DVL proteins, and has three α-helices, a β-hairpin arm, and two short β-strands. This domain enables interaction between DVL and DAAM1, thus activating the non-canonical pathway. This domain also has results that support claims that the DEP domain is what is responsible for targeting DVL proteins to the membrane upon Wnt signal stimulation. The DEP domain may also be essential for the assembly of functional signalosomes and for Wnt signal transduction to the nucleus.
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363
- Mlodzik M (2016). The Dishevelled Protein Family: Still Rather a Mystery After Over 20 Years of Molecular Studies. Vol. 117. pp. 75–91. doi:10.1016/bs.ctdb.2015.11.027. ISBN 9780128013823. PMC 4939608. PMID 26969973
NES and NLS regions
In addition to these conserved regions, DVL has both a NES and NLS, which regulate the cellular localization of DVL via movement between the nucleus and the cytoplasm. The NLS is between the PDZ and DEP domains, and the NES is between the DEP and C-terminus of DVL.
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363
There are three main types of DVL post-translational modification – phosphorylation, ubiquitination, and methylation. Phosphorylation is the most well-studied, and seems to act such that site-specific phosphorylation can bring about a wide variety of biological responses. Ubiquitination is the post-translational modification that has a role in regulating DVL degradation.
- Sharma M, Castro-Piedras I, Simmons GE, Pruitt K (July 2018). “Dishevelled: A masterful conductor of complex Wnt signals”. Cellular Signalling. 47: 52–64. doi:10.1016/j.cellsig.2018.03.004. PMC 6317740. PMID 29559363
The nerve secretes agrin, resulting in phosphorylation of the MuSK receptor.
It seems that the MuSK receptor recruits casein kinase 2, which is required for clustering.
- Cheusova T, Khan MA, Schubert SW, Gavin AC, Buchou T, Jacob G, et al. (July 2006). “Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction”. Genes & Development. 20 (13): 1800–1816. doi:10.1101/gad.375206. PMC 1522076. PMID 16818610.
A protein called rapsyn is then recruited to the primary MuSK scaffold, to induce the additional clustering of acetylcholine receptors (AChR). This is thought of as the secondary scaffold. A protein called Dok-7 has shown to be additionally required for the formation of the secondary scaffold; it is apparently recruited after MuSK phosphorylation and before acetylcholine receptors are clustered.
43 kDa receptor-associated protein of the synapse (rapsyn) is a protein that in humans is encoded by the RAPSN gene. This protein belongs to a family of proteins that are receptor associated proteins of the synapse. It contains a conserved cAMP-dependent protein kinase phosphorylation site. It is believed to play some role in anchoring or stabilizing the nicotinic acetylcholine receptor at synaptic sites. It may link the receptor to the underlying postsynaptic cytoskeleton, possibly by direct association with actin or spectrin. Two splice variants have been identified for this gene. In the neuromuscular junction there is a vital pathway that maintains synaptic structure and results in the aggregation and localization of the acetylcholine receptor (AChR) on the postsynaptic folds. This pathway consists of agrin, muscle-specific tyrosine kinase (MuSK protein), AChRs and the AChR-clustering protein rapsyn, encoded by RAPSN. Genetic mutations of the proteins in the neuromuscular junction are associated with Congenital myasthenic syndrome (CMS). Postsynaptic defects are the most frequent cause of CMS and often result in abnormalities in the acetylcholine receptor. The vast majority of mutations causing CMS are found in the AChR subunits and rapsyn genes.
The rapsyn protein interacts directly with the AChRs and plays a vital role in agrin-induced clustering of the AChR. Without rapsyn, functional synapses cannot be created as the folds do not form properly. Patients with CMS-related mutations of the rapsyn protein typically are either homozygous for N88K or heterozygous for N88K and a second mutation. The major effect of the mutation N88K in rapsyn is to reduce the stability of AChR clusters. The second mutation can be a determining factor in the severity of the disease.
Studies have shown that most patients with CMS that have rapsyn mutations carry the common mutation N88K on at least one allele. However, research has revealed that there is a small population of patients who do not carry the N88K mutation on either of their alleles, but instead have different mutations of the RAPSN gene on both of their alleles. Two novel missense mutations that have been found are R164C and L283P and the result is a decrease in co-clustering of AChR with raspyn. A third mutation is the intronic base alteration IVS1-15C>A and it causes abnormal splicing of RAPSN RNA. These results show that diagnostic screening for CMS mutations of the RAPSN gene cannot be based exclusively on the detection of N88K mutations Interestingly, patients who bear the burden of CMS due to these rapsyn mutations often demonstrate a remarkable response to anticholinesterase drugs like pyridostigmine. Moreover, the supplemental inclusion of 3,4 DAP, ephedrine, or albuterol often yields significant clinical improvement. RAPSN has been shown to interact with KHDRBS1.
- GRCh38: Ensembl release 89: ENSG00000165917 – Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000002104 – Ensembl, May 2017
- “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- “Mouse PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- Buckel A, Beeson D, James M, Vincent A (August 1996). “Cloning of cDNA encoding human rapsyn and mapping of the RAPSN gene locus to chromosome 11p11.2-p11.1”. Genomics. 35 (3): 613–616. doi:10.1006/geno.1996.0409. PMID 8812503.
- “Entrez Gene: RAPSN receptor-associated protein of the synapse”.
- Cossins J, Burke G, Maxwell S, Spearman H, Man S, Kuks J, et al. (October 2006). “Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations”. Brain. 129 (Pt 10): 2773–2783. doi:10.1093/brain/awl219. PMID 16945936.
- Müller JS, Baumeister SK, Rasic VM, Krause S, Todorovic S, Kugler K, et al. (October 2006). “Impaired receptor clustering in congenital myasthenic syndrome with novel RAPSN mutations”. Neurology. 67 (7): 1159–1164. doi:10.1212/01.wnl.0000233837.79459.40. PMID 16931511. S2CID 41593780.
- Liao X, Wang Y, Lai X, Wang S (February 2023). “The role of Rapsyn in neuromuscular junction and congenital myasthenic syndrome”. Biomolecules and Biomedicine. 23 (5): 772–784. doi:10.17305/bb.2022.8641. PMC 10494853. PMID 36815443. S2CID 257100080.
- Fung ET, Lanahan A, Worley P, Huganir RL (October 1998). “Identification of a Torpedo homolog of Sam68 that interacts with the synapse organizing protein rapsyn”. FEBS Letters. 437 (1–2): 29–33. doi:10.1016/S0014-5793(98)01151-X. PMID 9804166. S2CID 7842971.
Dok-7 is a non-catalytic cytoplasmic adaptor protein that is expressed specifically in muscle and is essential for the formation of neuromuscular synapses. Further, Dok-7 contains pleckstrin homology (PH) and phosphotyrosine-binding (PTB) domains that are critical for Dok-7 function. Finally, mutations in Dok-7 are commonly found in patients with limb-girdle congenital myasthenia.
Dok-7 regulates neuromuscular synapse formation by activating MuSK
The formation of neuromuscular synapses requires the muscle-specific receptor tyrosine kinase (MuSK). In mice genetically mutant for MuSK, acetylcholine receptors (AChRs) fail to cluster and motor neurons fail to differentiate. Because Dok-7 mutant mice are indistinguishable from MuSK mutant mice, these observations suggest Dok-7 might regulate MuSK activation. Indeed, Dok-7 binds phosphorylated MuSK and activates MuSK in purified protein preparations and in muscle in-vivo by transgenic overexpression. Furthermore, the nerve-derived organizing factor agrin fails to stimulate MuSK activation in muscle cells genetically null for Dok-7. Thus, Dok-7 is both necessary and sufficient for the activation of MuSK.
The requirement for MuSK in the formation of the NMJ was primarily demonstrated by mouse “knockout” studies. In mice which are deficient for either agrin or MuSK, the neuromuscular junction does not form.
Upon activation by its ligand agrin, MuSK signals via the proteins called Dok-7 and rapsyn, to induce “clustering” of acetylcholine receptors (AChR). Cell signaling downstream of MuSK requires Dok-7. Mice which lack this protein fail to develop endplates. Further, forced expression of Dok-7 induces the tyrosine phosphorylation, and thus the activation of MuSK. Dok-7 interacts with MuSK by way of protein “domain” called a “PTB domain.”
In addition to the AChR, MuSK, and Dok-7 other proteins are then gathered, to form the endplate to the neuromuscular junction. The nerve terminates onto the endplate, forming the neuromuscular junction—a structure which is required to transmit nerve impulses to the muscle, and thus initiating muscle contraction.
Homozygous mutation of Dok-7 is responsible for a form of congenital myasthenic syndrome (CMS) that is unique among disorders in this category because it affects muscles in the limbs and trunk but mostly spares the face, eyes, and functions of the mouth and pharynx (chewing, swallowing and speech). Salbutamol can be effective in relieving CMS symptoms attributable to Dok-7 mutations.
- Valenzuela DM, Stitt TN, DiStefano PS, et al. (Sep 1995). “Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury”. Neuron. 15 (3): 573–84. doi:10.1016/0896-6273(95)90146-9. PMID 7546737. S2CID 17575761.
- DeChiara TM, Bowen DC, Valenzuela DM, et al. (May 1996). “The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo”. Cell. 85 (4): 501–12. doi:10.1016/S0092-8674(00)81251-9. PMID 8653786. S2CID 17455481.
- Glass DJ, Bowen DC, Stitt TN, et al. (May 1996). “Agrin acts via a MuSK receptor complex”. Cell. 85 (4): 513–23. doi:10.1016/S0092-8674(00)81252-0. PMID 8653787. S2CID 14930468.
- Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A (Mar 2001). “Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies”. Nat. Med. 7 (3): 365–8. doi:10.1038/85520. PMID 11231638. S2CID 18641849.
- Strochlic L, Cartaud A, Cartaud J (Nov 2005). “The synaptic muscle-specific kinase (MuSK) complex: new partners, new functions” (PDF). BioEssays. 27 (11): 1129–35. doi:10.1002/bies.20305. PMID 16237673. S2CID 7386607.
- Okada K, Inoue A, Okada M, et al. (Jun 2006). “The muscle protein Dok-7 is essential for neuromuscular synaptogenesis”. Science. 312 (5781): 1802–5. Bibcode:2006Sci…312.1802O. doi:10.1126/science.1127142. PMID 16794080. S2CID 45730054.
- Palace J, Lashley D, Newsom-Davis J, et al. (Jun 2007). “Clinical features of the DOK7 neuromuscular junction synaptopathy”. Brain. 130 (Pt 6): 1507–15. CiteSeerX 10.1.1.547.7662. doi:10.1093/brain/awm072. PMID 17452375.
Structure
There are three potential heparan sulfate (HS) attachment sites within the primary structure of agrin, but it is thought that only two of these actually carry HS chains when the protein is expressed.
In fact, one study concluded that at least two attachment sites are necessary by inducing synthetic agents. Since agrin fragments induce acetylcholine receptor aggregation as well as phosphorylation of the MuSK receptor, researchers spliced them and found that the variant did not trigger phosphorylation. It has also been shown that the G3 domain of agrin is very plastic, meaning it can discriminate between binding partners for a better fit.
- PDB: 1PZ7; Stetefeld J, Alexandrescu AT, Maciejewski MW, Jenny M, Rathgeb-Szabo K, Schulthess T, et al. (March 2004). “Modulation of agrin function by alternative splicing and Ca2+ binding”. Structure. 12 (3): 503–515. doi:10.1016/j.str.2004.02.001. PMID 15016366.
Heparan sulfate glycosaminoglycans covalently linked to the agrin protein have been shown to play a role in the clustering of AChR. Interference in the correct formation of heparan sulfate through the addition of chlorate to skeletal muscle cell culture results in a decrease in the frequency of spontaneous acetylcholine receptor (AChR) clustering. It may be that rather than solely binding directly to the agrin protein core a number of components of the secondary scaffold may also interact with its heparan sulfate side-chains.
- McDonnell KM, Grow WA (2004). “Reduced glycosaminoglycan sulfation diminishes the agrin signal transduction pathway”. Developmental Neuroscience. 26 (1): 1–10. doi:10.1159/000080706. PMID 15509893. S2CID 42558266.
A role in the retention of anionic macromolecules within the vasculature has also been suggested for agrin-linked HS at the glomerular or alveolar basement membrane.
Functions
Agrin may play an important role in the basement membrane of the microvasculature as well as in synaptic plasticity. Also, agrin may be involved in blood–brain barrier (BBB) formation and/or function and it influences Aβ homeostasis.
- Donahue JE, Berzin TM, Rafii MS, Glass DJ, Yancopoulos GD, Fallon JR, Stopa EG (May 1999). “Agrin in Alzheimer’s disease: altered solubility and abnormal distribution within microvasculature and brain parenchyma”. Proceedings of the National Academy of Sciences of the United States of America. 96 (11): 6468–6472. Bibcode:1999PNAS…96.6468D. doi:10.1073/pnas.96.11.6468. PMC 26905. PMID 10339611.
- Wolburg H, Noell S, Wolburg-Buchholz K, Mack A, Fallier-Becker P (April 2009). “Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier”. The Neuroscientist. 15 (2): 180–193. doi:10.1177/1073858408329509. PMID 19307424. S2CID 25922007.
- Rauch SM, Huen K, Miller MC, Chaudry H, Lau M, Sanes JR, et al. (December 2011). “Changes in brain β-amyloid deposition and aquaporin 4 levels in response to altered agrin expression in mice”. Journal of Neuropathology and Experimental Neurology. 70 (12): 1124–1137. doi:10.1097/NEN.0b013e31823b0b12. PMC 3223604. PMID 22082664.
Research
Agrin is investigated in relation with osteoarthritis. In addition, by its ability to activate the Hippo signaling pathway, agrin is emerging as a key proteoglycan in the tumor microenvironment.
- Thorup A, Dell’Accio F, Eldridge SE (16 September 2020). “Regrowing knee cartilage: new animal studies show promise”. The Conversation. Retrieved 2020-10-12.
- Eldridge SE, Barawi A, Wang H, Roelofs AJ, Kaneva M, Guan Z, et al. (September 2020). “Agrin induces long-term osteochondral regeneration by supporting repair morphogenesis”. Science Translational Medicine. 12 (559): eaax9086. doi:10.1126/scitranslmed.aax9086. hdl:2164/15360. PMID 32878982. S2CID 221469142.
- Chakraborty S, Hong W (February 2018). “Linking Extracellular Matrix Agrin to the Hippo Pathway in Liver Cancer and Beyond”. Cancers. 10 (2): 45. doi:10.3390/cancers10020045. PMC 5836077. PMID 29415512.
Clinical significance
AGRN gene mutation leads to congenital myasthenic syndromes and myasthenia gravis.
- Gan S, Yang H, Xiao T, Pan Z, Wu L (October 2020). “AGRN Gene Mutation Leads to Congenital Myasthenia Syndromes: A Pediatric Case Report and Literature Review”. Neuropediatrics. 51 (5): 364–367. doi:10.1055/s-0040-1708534. PMID 32221959. S2CID 214694892.
- Ohkawara B, Shen X, Selcen D, Nazim M, Bril V, Tarnopolsky MA, et al. (April 2020). “Congenital myasthenic syndrome-associated agrin variants affect clustering of acetylcholine receptors in a domain-specific manner”. JCI Insight. 5 (7): 132023. doi:10.1172/jci.insight.132023. PMC 7205260. PMID 32271162.
- Xi J, Yan C, Liu WW, Qiao K, Lin J, Tian X, et al. (December 2017). “Novel SEA and LG2 Agrin mutations causing congenital Myasthenic syndrome”. Orphanet Journal of Rare Diseases. 12 (1): 182. doi:10.1186/s13023-017-0732-z. PMC 5735900. PMID 29258548.
- Zhang B, Shen C, Bealmear B, Ragheb S, Xiong WC, Lewis RA, et al. (2014-03-14). “Autoantibodies to agrin in myasthenia gravis patients”. PLOS ONE. 9 (3): e91816. Bibcode:2014PLoSO…991816Z. doi:10.1371/journal.pone.0091816. PMC 3954737. PMID 24632822.
- Yan M, Xing GL, Xiong WC, Mei L (February 2018). “Agrin and LRP4 antibodies as new biomarkers of myasthenia gravis”. Annals of the New York Academy of Sciences. 1413 (1): 126–135. Bibcode:2018NYASA1413..126Y. doi:10.1111/nyas.13573. PMID 29377176. S2CID 46757850
A recent genome-wide association study (GWAS) has found that genetic variations in AGRN are associated with late-onset sporadic Alzheimer’s disease (LOAD). These genetic variations alter β-amyloid homeostasis contributing to its accumulation and plaque formation.
- Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, et al. (September 2021). “A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease”. Nature Genetics. 53 (9): 1276–1282. doi:10.1038/s41588-021-00921-z. hdl:1871.1/61f01aa9-6dc7-4213-be2a-d3fe622db488. PMC 10243600. PMID 34493870. S2CID 237442349.
- Rahman MM, Lendel C (August 2021). “Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology”. Molecular Neurodegeneration. 16 (1): 59. doi:10.1186/s13024-021-00465-0. PMC 8400902. PMID 34454574.
References
- GRCh38: Ensembl release 89: ENSG00000188157 – Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000041936 – Ensembl, May 2017
- “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- “Mouse PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- Rupp F, Payan DG, Magill-Solc C, Cowan DM, Scheller RH (May 1991). “Structure and expression of a rat agrin”. Neuron. 6 (5): 811–823. doi:10.1016/0896-6273(91)90177-2. PMID 1851019. S2CID 44440186.
- Kröger S, Schröder JE (October 2002). “Agrin in the developing CNS: new roles for a synapse organizer”. News in Physiological Sciences. 17 (5): 207–212. doi:10.1152/nips.01390.2002. PMID 12270958. S2CID 2988918.
- Groffen AJ, Buskens CA, van Kuppevelt TH, Veerkamp JH, Monnens LA, van den Heuvel LP (May 1998). “Primary structure and high expression of human agrin in basement membranes of adult lung and kidney”. European Journal of Biochemistry. 254 (1): 123–128. doi:10.1046/j.1432-1327.1998.2540123.x. PMID 9652404.
- Tsen G, Halfter W, Kröger S, Cole GJ (February 1995). “Agrin is a heparan sulfate proteoglycan”. The Journal of Biological Chemistry. 270 (7): 3392–3399. doi:10.1074/jbc.270.7.3392. PMID 7852425.
- Groffen AJ, Ruegg MA, Dijkman H, van de Velden TJ, Buskens CA, van den Born J, et al. (January 1998). “Agrin is a major heparan sulfate proteoglycan in the human glomerular basement membrane”. The Journal of Histochemistry and Cytochemistry. 46 (1): 19–27. doi:10.1177/002215549804600104. PMID 9405491. S2CID 24353753.
- Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR, Thomas S, LeBeau MM, Fernald AA, Copeland NG, Jenkins NA, Burden SJ, Glass DJ, Yancopoulos GD (Sep 1995). “Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury”. Neuron. 15 (3): 573–584. doi:10.1016/0896-6273(95)90146-9. PMID 7546737.
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD (May 1996). “The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo”. Cell. 85 (4): 501–512. doi:10.1016/s0092-8674(00)81251-9. PMID 8653786.
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD (May 1996). “Agrin acts via a MuSK receptor complex”. Cell. 85 (4): 513–523. doi:10.1016/s0092-8674(00)81252-0. PMID 8653787.
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (May 1996). “Defective neuromuscular synaptogenesis in agrin-deficient mutant mice”. Cell. 85 (4): 525–535. doi:10.1016/s0092-8674(00)81253-2. PMID 8653788.
- Magill C, Reist NE, Fallon JR, Nitkin RM, Wallace BG, McMahan UJ (1987). “Chapter 32 Agrin”. Neural Regeneration. Progress in Brain Research. Vol. 71. pp. 391–396. doi:10.1016/S0079-6123(08)61840-3. ISBN 978-0-444-80814-1. PMID 3035610.
- Sanes JR, Lichtman JW (November 2001). “Induction, assembly, maturation and maintenance of a postsynaptic apparatus”. Nature Reviews. Neuroscience. 2 (11): 791–805. doi:10.1038/35097557. PMID 11715056. S2CID 52802445.
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, et al. (May 1996). “Agrin acts via a MuSK receptor complex”. Cell. 85 (4): 513–523. doi:10.1016/S0092-8674(00)81252-0. PMID 8653787. S2CID 14930468.
- Sanes JR, Apel ED, Gautam M, Glass D, Grady RM, Martin PT, et al. (May 1998). “Agrin receptors at the skeletal neuromuscular junction”. Annals of the New York Academy of Sciences. 841 (1): 1–13. Bibcode:1998NYASA.841….1S. doi:10.1111/j.1749-6632.1998.tb10905.x. PMID 9668217. S2CID 20097480.
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (May 1996). “Defective neuromuscular synaptogenesis in agrin-deficient mutant mice”. Cell. 85 (4): 525–535. doi:10.1016/S0092-8674(00)81253-2. PMID 8653788. S2CID 12517490.
- Cheusova T, Khan MA, Schubert SW, Gavin AC, Buchou T, Jacob G, et al. (July 2006). “Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction”. Genes & Development. 20 (13): 1800–1816. doi:10.1101/gad.375206. PMC 1522076. PMID 16818610.
- PDB: 1PZ7; Stetefeld J, Alexandrescu AT, Maciejewski MW, Jenny M, Rathgeb-Szabo K, Schulthess T, et al. (March 2004). “Modulation of agrin function by alternative splicing and Ca2+ binding”. Structure. 12 (3): 503–515. doi:10.1016/j.str.2004.02.001. PMID 15016366.
- McDonnell KM, Grow WA (2004). “Reduced glycosaminoglycan sulfation diminishes the agrin signal transduction pathway”. Developmental Neuroscience. 26 (1): 1–10. doi:10.1159/000080706. PMID 15509893. S2CID 42558266.
- Donahue JE, Berzin TM, Rafii MS, Glass DJ, Yancopoulos GD, Fallon JR, Stopa EG (May 1999). “Agrin in Alzheimer’s disease: altered solubility and abnormal distribution within microvasculature and brain parenchyma”. Proceedings of the National Academy of Sciences of the United States of America. 96 (11): 6468–6472. Bibcode:1999PNAS…96.6468D. doi:10.1073/pnas.96.11.6468. PMC 26905. PMID 10339611.
- Wolburg H, Noell S, Wolburg-Buchholz K, Mack A, Fallier-Becker P (April 2009). “Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier”. The Neuroscientist. 15 (2): 180–193. doi:10.1177/1073858408329509. PMID 19307424. S2CID 25922007.
- Rauch SM, Huen K, Miller MC, Chaudry H, Lau M, Sanes JR, et al. (December 2011). “Changes in brain β-amyloid deposition and aquaporin 4 levels in response to altered agrin expression in mice”. Journal of Neuropathology and Experimental Neurology. 70 (12): 1124–1137. doi:10.1097/NEN.0b013e31823b0b12. PMC 3223604. PMID 22082664.
- Thorup A, Dell’Accio F, Eldridge SE (16 September 2020). “Regrowing knee cartilage: new animal studies show promise”. The Conversation. Retrieved 2020-10-12.
- Eldridge SE, Barawi A, Wang H, Roelofs AJ, Kaneva M, Guan Z, et al. (September 2020). “Agrin induces long-term osteochondral regeneration by supporting repair morphogenesis”. Science Translational Medicine. 12 (559): eaax9086. doi:10.1126/scitranslmed.aax9086. hdl:2164/15360. PMID 32878982. S2CID 221469142.
- Chakraborty S, Hong W (February 2018). “Linking Extracellular Matrix Agrin to the Hippo Pathway in Liver Cancer and Beyond”. Cancers. 10 (2): 45. doi:10.3390/cancers10020045. PMC 5836077. PMID 29415512.
- Gan S, Yang H, Xiao T, Pan Z, Wu L (October 2020). “AGRN Gene Mutation Leads to Congenital Myasthenia Syndromes: A Pediatric Case Report and Literature Review”. Neuropediatrics. 51 (5): 364–367. doi:10.1055/s-0040-1708534. PMID 32221959. S2CID 214694892.
- Ohkawara B, Shen X, Selcen D, Nazim M, Bril V, Tarnopolsky MA, et al. (April 2020). “Congenital myasthenic syndrome-associated agrin variants affect clustering of acetylcholine receptors in a domain-specific manner”. JCI Insight. 5 (7): 132023. doi:10.1172/jci.insight.132023. PMC 7205260. PMID 32271162.
- Xi J, Yan C, Liu WW, Qiao K, Lin J, Tian X, et al. (December 2017). “Novel SEA and LG2 Agrin mutations causing congenital Myasthenic syndrome”. Orphanet Journal of Rare Diseases. 12 (1): 182. doi:10.1186/s13023-017-0732-z. PMC 5735900. PMID 29258548.
- Zhang B, Shen C, Bealmear B, Ragheb S, Xiong WC, Lewis RA, et al. (2014-03-14). “Autoantibodies to agrin in myasthenia gravis patients”. PLOS ONE. 9 (3): e91816. Bibcode:2014PLoSO…991816Z. doi:10.1371/journal.pone.0091816. PMC 3954737. PMID 24632822.
- Yan M, Xing GL, Xiong WC, Mei L (February 2018). “Agrin and LRP4 antibodies as new biomarkers of myasthenia gravis”. Annals of the New York Academy of Sciences. 1413 (1): 126–135. Bibcode:2018NYASA1413..126Y. doi:10.1111/nyas.13573. PMID 29377176. S2CID 46757850.
- Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, et al. (September 2021). “A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease”. Nature Genetics. 53 (9): 1276–1282. doi:10.1038/s41588-021-00921-z. hdl:1871.1/61f01aa9-6dc7-4213-be2a-d3fe622db488. PMC 10243600. PMID 34493870. S2CID 237442349.
- Rahman MM, Lendel C (August 2021). “Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology”. Molecular Neurodegeneration. 16 (1): 59. doi:10.1186/s13024-021-00465-0. PMC 8400902. PMID 34454574.
Further reading
- Gros K, Matkovič U, Parato G, Miš K, Luin E, Bernareggi A, et al. (October 2022). “Neuronal Agrin Promotes Proliferation of Primary Human Myoblasts in an Age-Dependent Manner”. International Journal of Molecular Sciences. 23 (19): 11784. doi:10.3390/ijms231911784. PMC 9570459. PMID 36233091.
- Kesari S, Lasner TM, Balsara KR, Randazzo BP, Lee VM, Trojanowski JQ, Fraser NW (March 1998). “A neuroattenuated ICP34.5-deficient herpes simplex virus type 1 replicates in ependymal cells of the murine central nervous system”. The Journal of General Virology. 79 ( Pt 3) (3): 525–36. doi:10.1099/0022-1317-79-3-525. PMID 9519831.
- dde Souza Ramos JT, Ferrari FS, Andrade MF, de Melo CS, Boas PJ, Costa NA, et al. (February 2022). “Association between frailty and C-terminal agrin fragment with 3-month mortality following ST-elevation myocardial infarction”. Experimental Gerontology. 158: 111658. doi:10.1016/j.exger.2021.111658. PMID 34920013. S2CID 245149341.
- Zieliński AE (1996). “Specific immunotherapy in pollinosis: I. Valuation of certain cytoimmunological indicators over the course of four years of immunotherapy in pollinosis”. Journal of Investigational Allergology & Clinical Immunology. 6 (5): 307–14. PMID 8959542.
- Pizzuti A, Amati F, Calabrese G, et al. (1997). “cDNA characterization and chromosomal mapping of two human homologues of the Drosophila dishevelled polarity gene”. Hum. Mol. Genet. 5 (7): 953–8. doi:10.1093/hmg/5.7.953. PMID 8817329.
- Klingensmith J, Yang Y, Axelrod JD, et al. (1997). “Conservation of dishevelled structure and function between flies and mice: isolation and characterization of Dvl2”. Mech. Dev. 58 (1–2): 15–26. doi:10.1016/S0925-4773(96)00549-7. PMID 8887313. S2CID 16004832.
- Semënov MV, Snyder M (1997). “Human dishevelled genes constitute a DHR-containing multigene family”. Genomics. 42 (2): 302–10. doi:10.1006/geno.1997.4713. PMID 9192851.
- Strovel ET, Wu D, Sussman DJ (2000). “Protein phosphatase 2Calpha dephosphorylates axin and activates LEF-1-dependent transcription”. J. Biol. Chem. 275 (4): 2399–403. doi:10.1074/jbc.275.4.2399. PMID 10644691.
- Song DH, Sussman DJ, Seldin DC (2000). “Endogenous protein kinase CK2 participates in Wnt signaling in mammary epithelial cells”. J. Biol. Chem. 275 (31): 23790–7. doi:10.1074/jbc.M909107199. PMID 10806215.
- Zhang Y, Neo SY, Han J, Lin SC (2000). “Dimerization choices control the ability of axin and dishevelled to activate c-Jun N-terminal kinase/stress-activated protein kinase”. J. Biol. Chem. 275 (32): 25008–14. doi:10.1074/jbc.M002491200. PMID 10829020.
- Chen W, Hu LA, Semenov MV, et al. (2002). “β-Arrestin1 modulates lymphoid enhancer factor transcriptional activity through interaction with phosphorylated dishevelled proteins”. Proc. Natl. Acad. Sci. U.S.A. 98 (26): 14889–94. Bibcode:2001PNAS…9814889C. doi:10.1073/pnas.211572798. PMC 64954. PMID 11742073.
- Habas R, Kato Y, He X (2002). “Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1”. Cell. 107 (7): 843–54. doi:10.1016/S0092-8674(01)00614-6. PMID 11779461. S2CID 16894557.
- Strausberg RL, Feingold EA, Grouse LH, et al. (2003). “Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences”. Proc. Natl. Acad. Sci. U.S.A. 99 (26): 16899–903. Bibcode:2002PNAS…9916899M. doi:10.1073/pnas.242603899. PMC 139241. PMID 12477932.
- Habas R, Dawid IB, He X (2003). “Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation”. Genes Dev. 17 (2): 295–309. doi:10.1101/gad.1022203. PMC 195976. PMID 12533515.
- Chen W, ten Berge D, Brown J, et al. (2003). “Dishevelled 2 recruits beta-arrestin 2 to mediate Wnt5A-stimulated endocytosis of Frizzled 4”. Science. 301 (5638): 1391–4. Bibcode:2003Sci…301.1391C. doi:10.1126/science.1082808. PMID 12958364. S2CID 34975427.
- Wong CK, Luo W, Deng Y, et al. (2004). “The DIX domain protein coiled-coil-DIX1 inhibits c-Jun N-terminal kinase activation by Axin and dishevelled through distinct mechanisms”. J. Biol. Chem. 279 (38): 39366–73. doi:10.1074/jbc.M404598200. PMID 15262978.
- Torban E, Wang HJ, Groulx N, Gros P (2005). “Independent mutations in mouse Vangl2 that cause neural tube defects in looptail mice impair interaction with members of the Dishevelled family”. J. Biol. Chem. 279 (50): 52703–13. doi:10.1074/jbc.M408675200. PMID 15456783.
- Gerhard DS, Wagner L, Feingold EA, et al. (2004). “The Status, Quality, and Expansion of the NIH Full-Length cDNA Project: The Mammalian Gene Collection (MGC)”. Genome Res. 14 (10B): 2121–7. doi:10.1101/gr.2596504. PMC 528928. PMID 15489334.
- Rual JF, Venkatesan K, Hao T, et al. (2005). “Towards a proteome-scale map of the human protein-protein interaction network”. Nature. 437 (7062): 1173–8. Bibcode:2005Natur.437.1173R. doi:10.1038/nature04209. PMID 16189514. S2CID 4427026.
- Zhang L, Gao X, Wen J, et al. (2006). “Dapper 1 antagonizes Wnt signaling by promoting dishevelled degradation”. J. Biol. Chem. 281 (13): 8607–12. doi:10.1074/jbc.M600274200. PMID 16446366.
- Lim J, Hao T, Shaw C, et al. (2006). “A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration”. Cell. 125 (4): 801–14. doi:10.1016/j.cell.2006.03.032. PMID 16713569. S2CID 13709685.
- Apel ED, Roberds SL, Campbell KP, Merlie JP (July 1995). “Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex”. Neuron. 15 (1): 115–126. doi:10.1016/0896-6273(95)90069-1. PMID 7619516. S2CID 589282.
- Apel ED, Glass DJ, Moscoso LM, Yancopoulos GD, Sanes JR (April 1997). “Rapsyn is required for MuSK signaling and recruits synaptic components to a MuSK-containing scaffold”. Neuron. 18 (4): 623–635. doi:10.1016/S0896-6273(00)80303-7. PMID 9136771. S2CID 18020725.
- Yang SH, Armson PF, Cha J, Phillips WD (1997). “Clustering of GABAA receptors by rapsyn/43kD protein in vitro”. Molecular and Cellular Neurosciences. 8 (6): 430–438. doi:10.1006/mcne.1997.0597. PMID 9143560. S2CID 140208903.
- Ramarao MK, Cohen JB (March 1998). “Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn”. Proceedings of the National Academy of Sciences of the United States of America. 95 (7): 4007–4012. Bibcode:1998PNAS…95.4007R. doi:10.1073/pnas.95.7.4007. PMC 19953. PMID 9520483.
- Fung ET, Lanahan A, Worley P, Huganir RL (October 1998). “Identification of a Torpedo homolog of Sam68 that interacts with the synapse organizing protein rapsyn”. FEBS Letters. 437 (1–2): 29–33. doi:10.1016/S0014-5793(98)01151-X. PMID 9804166. S2CID 7842971.
- Qian X, Riccio A, Zhang Y, Ginty DD (November 1998). “Identification and characterization of novel substrates of Trk receptors in developing neurons”. Neuron. 21 (5): 1017–1029. doi:10.1016/S0896-6273(00)80620-0. PMID 9856458. S2CID 12354383.
- Zhou H, Glass DJ, Yancopoulos GD, Sanes JR (September 1999). “Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations”. The Journal of Cell Biology. 146 (5): 1133–1146. doi:10.1083/jcb.146.5.1133. PMC 2169478. PMID 10477765.
- Han H, Noakes PG, Phillips WD (September 1999). “Overexpression of rapsyn inhibits agrin-induced acetylcholine receptor clustering in muscle cells”. Journal of Neurocytology. 28 (9): 763–775. doi:10.1023/A:1007098406748. PMID 10859577. S2CID 36518335.
- Ramarao MK, Bianchetta MJ, Lanken J, Cohen JB (March 2001). “Role of rapsyn tetratricopeptide repeat and coiled-coil domains in self-association and nicotinic acetylcholine receptor clustering”. The Journal of Biological Chemistry. 276 (10): 7475–7483. doi:10.1074/jbc.M009888200. PMID 11087759.
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF (April 2001). “Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse”. Nature. 410 (6832): 1057–1064. Bibcode:2001Natur.410.1057L. doi:10.1038/35074025. PMID 11323662. S2CID 4372222.
- Bartoli M, Ramarao MK, Cohen JB (July 2001). “Interactions of the rapsyn RING-H2 domain with dystroglycan”. The Journal of Biological Chemistry. 276 (27): 24911–24917. doi:10.1074/jbc.M103258200. PMID 11342559.
- Ohno K, Engel AG, Shen XM, Selcen D, Brengman J, Harper CM, et al. (April 2002). “Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome”. American Journal of Human Genetics. 70 (4): 875–885. doi:10.1086/339465. PMC 379116. PMID 11791205.
- Marchand S, Devillers-Thiéry A, Pons S, Changeux JP, Cartaud J (October 2002). “Rapsyn escorts the nicotinic acetylcholine receptor along the exocytic pathway via association with lipid rafts”. The Journal of Neuroscience. 22 (20): 8891–8901. doi:10.1523/JNEUROSCI.22-20-08891.2002. PMC 6757681. PMID 12388596.
- Huebsch KA, Maimone MM (February 2003). “Rapsyn-mediated clustering of acetylcholine receptor subunits requires the major cytoplasmic loop of the receptor subunits”. Journal of Neurobiology. 54 (3): 486–501. doi:10.1002/neu.10177. PMID 12532399.
- Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG (April 2003). “E-box mutations in the RAPSN promoter region in eight cases with congenital myasthenic syndrome”. Human Molecular Genetics. 12 (7): 739–748. doi:10.1093/hmg/ddg089. PMID 12651869.
- Dunne V, Maselli RA (2003). “Identification of pathogenic mutations in the human rapsyn gene”. Journal of Human Genetics. 48 (4): 204–207. doi:10.1007/s10038-003-0005-7. PMID 12730725.
- Müller JS, Mildner G, Müller-Felber W, Schara U, Krampfl K, Petersen B, et al. (June 2003). “Rapsyn N88K is a frequent cause of congenital myasthenic syndromes in European patients”. Neurology. 60 (11): 1805–1810. doi:10.1212/01.wnl.0000072262.14931.80. PMID 12796535. S2CID 30030861.
- Richard P, Gaudon K, Andreux F, Yasaki E, Prioleau C, Bauché S, et al. (June 2003). “Possible founder effect of rapsyn N88K mutation and identification of novel rapsyn mutations in congenital myasthenic syndromes”. Journal of Medical Genetics. 40 (6): 81e–81. doi:10.1136/jmg.40.6.e81. PMC 1735489. PMID 12807980.
External links
- Human AGRN genome location and AGRN gene details page in the UCSC Genome Browser.
| Protein kinases: tyrosine kinases (EC 2.7.10) |
|---|
Leave a Reply