Isoleucine, Tryptophol, Sleeping Sickness, The Disulfiram Effect and One Trick Hypnotists From Hell
Isoleucine (symbol Ile or I) is an α-amino acid that is used in the biosynthesis of proteins. It contains an α-amino group (which is in the protonated −NH+3 form under biological conditions), an α-carboxylic acid group (which is in the deprotonated −COO− form under biological conditions), and a hydrocarbon side chain with a branch (a central carbon atom bound to three other carbon atoms). It is classified as a non-polar, uncharged (at physiological pH), branched-chain, aliphatic amino acid. It is essential in humans, meaning the body cannot synthesize it. Essential amino acids are necessary in the human diet. In plants isoleucine can be synthesized from threonine and methionine. In plants and bacteria, isoleucine is synthesized from pyruvate employing leucine biosynthesis enzymes. It is encoded by the codons AUU, AUC, and AUA.
- “IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature and symbolism for amino acids and peptides. Recommendations 1983”. The Biochemical Journal. 219 (2): 345–373. April 1984. doi:10.1042/bj2190345. PMC 1153490. PMID 6743224.
- Joshi V, Joung JG, Fei Z, Jander G (October 2010). “Interdependence of threonine, methionine and isoleucine metabolism in plants: accumulation and transcriptional regulation under abiotic stress”. Amino Acids. 39 (4): 933–947. doi:10.1007/s00726-010-0505-7. PMID 20186554. S2CID 22641155.
- Kisumi M, Komatsubara S, Chibata I (July 1977). “Pathway for isoleucine formation form pyruvate by leucine biosynthetic enzymes in leucine-accumulating isoleucine revertants of Serratia marcescens”. Journal of Biochemistry. 82 (1): 95–103. doi:10.1093/oxfordjournals.jbchem.a131698. PMID 142769.

Metabolism
Biosynthesis
In plants and microorganisms, isoleucine is synthesized from pyruvate and alpha-ketobutyrate. This pathway is not present in humans. Enzymes involved in this biosynthesis include:
- Acetolactate synthase (also known as acetohydroxy acid synthase)
- Acetohydroxy acid isomeroreductase
- Dihydroxyacid dehydratase
- Valine aminotransferase
Catabolism
Isoleucine is both a glucogenic and a ketogenic amino acid. After transamination with alpha-ketoglutarate, the carbon skeleton is oxidised and split into propionyl-CoA and acetyl-CoA. Propionyl-CoA is converted into succinyl-CoA, a TCA cycle intermediate which can be converted into oxaloacetate for gluconeogenesis (hence glucogenic). In mammals acetyl-CoA cannot be converted to carbohydrate but can be either fed into the TCA cycle by condensing with oxaloacetate to form citrate or used in the synthesis of ketone bodies (hence ketogenic) or fatty acids.
- Lehninger AL, Nelson DL, Cox MM (2000). Lehninger principles of biochemistry (3rd ed.). New York: Worth Publishers. ISBN 1-57259-153-6. OCLC 42619569.
- Rajendram R, Preedy VR, Patel VB (2015). Branched chain amino acids in clinical nutrition. Vol. 1. New York, New York: Humana. ISBN 978-1-4939-1923-9. OCLC 898999904.
Metabolic diseases
The degradation of isoleucine is impaired in the following metabolic diseases:
- Combined malonic and methylmalonic aciduria (CMAMMA)
- Maple syrup urine disease (MSUD)
- Methylmalonic acidemia
- Propionic acidemia
Insulin resistance
Isoleucine, like other branched-chain amino acids, is associated with insulin resistance: higher levels of isoleucine are observed in the blood of diabetic mice, rats, and humans. In diet-induced obese and insulin resistant mice, a diet with decreased levels of isoleucine (with or without the other branched-chain amino acids) results in reduced adiposity and improved insulin sensitivity. Reduced dietary levels of isoleucine are required for the beneficial metabolic effects of a low protein diet. In humans, a protein restricted diet lowers blood levels of isoleucine and decreases fasting blood glucose levels. Mice fed a low isoleucine diet are leaner, live longer, and are less frail. In humans, higher dietary levels of isoleucine are associated with greater body mass index.
- Lynch CJ, Adams SH (December 2014). “Branched-chain amino acids in metabolic signalling and insulin resistance”. Nature Reviews. Endocrinology. 10 (12): 723–736. doi:10.1038/nrendo.2014.171. PMC 4424797. PMID 25287287.
- Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. (February 2018). “Restoration of metabolic health by decreased consumption of branched-chain amino acids”. The Journal of Physiology. 596 (4): 623–645. doi:10.1113/JP275075. PMC 5813603. PMID 29266268.
- Yu D, Richardson NE, Green CL, Spicer AB, Murphy ME, Flores V, et al. (May 2021). “The adverse metabolic effects of branched-chain amino acids are mediated by isoleucine and valine”. Cell Metabolism. 33 (5): 905–922.e6. doi:10.1016/j.cmet.2021.03.025. PMC 8102360. PMID 33887198.
- Fontana L, Cummings NE, Arriola Apelo SI, Neuman JC, Kasza I, Schmidt BA, et al. (July 2016). “Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health”. Cell Reports. 16 (2): 520–530. doi:10.1016/j.celrep.2016.05.092. PMC 4947548. PMID 27346343.
- Green CL, Trautman ME, Chaiyakul K, Jain R, Alam YH, Babygirija R, et al. (November 2023). “Dietary restriction of isoleucine increases healthspan and lifespan of genetically heterogeneous mice”. Cell Metabolism. 35 (11): 1976–1995.e6. doi:10.1016/j.cmet.2023.10.005. PMC 10655617. PMID 37939658.
Functions and requirement
The Food and Nutrition Board (FNB) of the U.S. Institute of Medicine has set Recommended Dietary Allowances (RDAs) for essential amino acids in 2002. For adults 19 years and older, 19 mg of isoleucine/kg body weight is required daily.
- Institute of Medicine. Panel on Macronutrients, Institute of Medicine. Standing Committee on the Scientific Evaluation of Dietary Reference Intakes (2005). Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. Washington, D.C.: National Academies Press. ISBN 0-309-08537-3. OCLC 57373786.
Beside its biological role as a nutrient, isoleucine also participates in regulation of glucose metabolism. Isoleucine is an essential component of many proteins. As an essential amino acid, isoleucine must be ingested or protein production in the cell will be disrupted. Fetal hemoglobin is one of the many proteins that require isoleucine. Isoleucine is present in the gamma chain of fetal hemoglobin and must be present for the protein to form.
- Rajendram R, Preedy VR, Patel VB (2015). Branched chain amino acids in clinical nutrition. Vol. 1. New York, New York: Humana. ISBN 978-1-4939-1923-9. OCLC 898999904.
- Honig GR (November 1967). “Inhibition of synthesis of fetal hemoglobin by an isoleucine analogue”. The Journal of Clinical Investigation. 46 (11): 1778–1784. doi:10.1172/JCI105668. PMC 292928. PMID 4964832.
Genetic diseases can change the consumption requirements of isoleucine. Amino acids cannot be stored in the body. Buildup of excess amino acids will cause a buildup of toxic molecules so, humans have many pathways to degrade each amino acid when the need for protein synthesis has been met. Mutations in isoleucine-degrading enzymes can lead to dangerous buildup of isoleucine and it’s toxic derivative. One example is maple syrup urine disease (MSUD), a disorder that leaves people unable to breakdown isoleucine, valine, and leucine. People with MSUD manage their disease by a reduced intake of all three of those amino acids alongside drugs that help excrete built-up toxins.
- Korman SH (December 2006). “Inborn errors of isoleucine degradation: a review”. Molecular Genetics and Metabolism. 89 (4): 289–299. doi:10.1016/j.ymgme.2006.07.010. PMID 16950638.
- Hassan SA, Gupta V (2023). “Maple Syrup Urine Disease”. StatPearls. Treasure Island (FL): StatPearls Publishing. PMID 32491705. Retrieved 2023-04-16.
- Brunetti-Pierri N, Lanpher B, Erez A, Ananieva EA, Islam M, Marini JC, et al. (February 2011). “Phenylbutyrate therapy for maple syrup urine disease”. Human Molecular Genetics. 20 (4): 631–640. doi:10.1093/hmg/ddq507. PMC 3024040. PMID 21098507
Many animals and plants are dietary sources of isoleucine as a component of proteins. Foods that have high amounts of isoleucine include eggs, soy protein, seaweed, turkey, chicken, lamb, cheese, and fish.
- Rajendram R, Preedy VR, Patel VB (2015). Branched chain amino acids in clinical nutrition. Vol. 1. New York, New York: Humana. ISBN 978-1-4939-1923-9. OCLC 898999904.
Synthesis
Routes to isoleucine are numerous. One common multistep procedure starts from 2-bromobutane and diethylmalonate. Synthetic isoleucine was first reported in 1905 by French chemists Bouveault and Locquin.
- Marvel CS (1941). Bachmann WE, Holmes DW (eds.). “dl-Isoleucine”. Organic Syntheses. 21: 60. doi:10.15227/orgsyn.021.0060. ISSN 0078-6209.
- Bouvealt L, Locquin R (1905). “Sur la synthése d’une nouvelle leucine”. Compt. Rend. (141): 115–117.
See also
Discovery
Felix Ehrlich (1877 – 1942) was a German chemist and biochemist who studied in Berlin and Munich. After receiving his doctorate in 1900, he worked at the Institute of Sugar Industry in Berlin. In 1906 he obtained his diploma in chemistry. From 1909 he worked as professor in Breslau, and later as director of the Institute on Biotechnology and Agriculture.

Ehrlich discovered the amino acid isoleucine in hemoglobin in 1903, developed a process for resolving racemic amino acids in 1906, described the formation of fusel oils by fermentation, amino acid during alcoholic fermentation in 1905 and worked on the structure of pectins. He was the recipient of the 1931 Emil Fischer Medal. That guy, again.
Another page says Ehrlich discovered isoleucine while studying the composition of beet-sugar molasses 1903. In 1907 Ehrlich carried out further studies on fibrin, egg albumin, gluten, and beef muscle in 1907. These studies verified the natural composition of isoleucine. Ehrlich published his own synthesis of isoleucine in 1908.
- Vickery HB, Schmidt CL (October 1931). “The History of the Discovery of the Amino Acids”. Chemical Reviews. 9 (2): 169–318. doi:10.1021/cr60033a001. ISSN 0009-2665.
- Ehrlich F (1908). “Über eine Synthese des Isoleucins”. Chemische Berichte. 41 (1): 1453–1458. doi:10.1002/cber.190804101266. ISSN 0365-9496.
Ehrlich demonstrated that yeast attacks the natural amino acids essentially by splitting off carbon dioxide and replacing the amino group with hydroxyl. By this reaction, the tryptophan gives rise to tryptophol.
- Olbrich, Hubert (2004). Zucker-Museum 1904 bis 2004: Beiträge zum Jubiläumsjahr, Volume 3. Berlin: Technische Universität, Universitätsbibliothek. p. 600.
- Hartmann, Guido. “Felix Ehrlich”. Deutsche Biographie. Retrieved November 3, 2020.
- Kranich, Kai (2018). Die “Bollwerk-Ingenieure”: Technikwissenschaft in Breslau 1900–1945. Paderborn: Verlag Ferdinand Schöningh.
- A synthesis of tryptophol. Richard W. Jackson, The Journal of Biological Chemistry, vol. 88, number 3, pp. 659–662
- Jüdisches biographisches Lexikon: eine Sammlung von bedeutenden … by Hans Morgenstern, P. 190 on Google books
Tryptophol is an aromatic alcohol that induces sleep in humans. It is found in wine as a secondary product of ethanol fermentation. It was first described by Felix Ehrlich in 1912. It is also produced by the trypanosomal parasite in sleeping sickness.

Sleeping Sickness has been present in Africa for thousands of years. Because of a lack of travel between indigenous people, sleeping sickness in humans had been limited to isolated pockets. This changed after Arab slave traders entered central Africa from the east, following the Congo River, bringing parasites along. Gambian sleeping sickness travelled up the Congo River, and then further east.
- Steverding D (February 2008). “The history of African trypanosomiasis”. Parasites & Vectors. 1 (1): 3. doi:10.1186/1756-3305-1-3. PMC 2270819. PMID 18275594.
- Strong RP (1944). Stitt’s Diagnosis, Prevention and Treatment of Tropical Diseases (Seventh ed.). York, PA: The Blakiston company. p. 165.
An Arab writer of the 14th century left the following description in the case of a sultan of the Mali Kingdom:
“His end was to be overtaken by the sleeping sickness (illat an-nawm) which is a disease that frequently befalls the inhabitants of these countries especially their chieftains. Sleep overtakes one of them in such a manner that it is hardly possible to awake him.”
Strong RP (1944). Stitt’s Diagnosis, Prevention and Treatment of Tropical Diseases (Seventh ed.). York, PA: The Blakiston company. p. 165
The British naval surgeon John Atkins described the disease on his return home from West Africa in 1734:
The Sleepy Distemper…gives no other previous Notice, than a want of Appetite 2 or 3 days before; their sleeps are sound, and Sense and Feeling very little; for pulling, drubbing or whipping will scarce stir up Sense and Power enough to move; and the Moment you cease beating the smart is forgot, and down they fall again into a state of Insensibility, drivling constantly from the Mouth as in deep salivation; breathe slowly, but not unequally nor snort. Young people are more subject to it than the old; and the Judgement generally pronounced is Death, the Prognostik seldom failing. If now and then one of them recovers, he certainly loses the little Reason he had, and turns Ideot…
Strong RP (1944). Stitt’s Diagnosis, Prevention and Treatment of Tropical Diseases (Seventh ed.). York, PA: The Blakiston company. p. 165.
French naval surgeon Marie-Théophile Griffon du Bellay treated and described cases while stationed aboard the hospital ship Caravane in Gabon in the late 1860s. In 1901, a devastating epidemic erupted in Uganda, killing more than 250,000 people, including about two-thirds of the population in the affected lakeshore areas. According to The Cambridge History of Africa, “It has been estimated that up to half the people died of sleeping-sickness and smallpox in the lands on either bank of the lower river Congo.”
- “Médecin”. ecole.nav.traditions.free.fr.
- Fèvre EM, Coleman PG, Welburn SC, Maudlin I (April 2004). “Reanalyzing the 1900-1920 sleeping sickness epidemic in Uganda”. Emerging Infectious Diseases. 10 (4): 567–73. doi:10.3201/eid1004.020626. PMID 15200843.
- Fage JD (5 September 1985). The Cambridge History of Africa: From the earliest times to c. 500 BC. Cambridge University Press. p. 748. ISBN 978-0-521-22803-9. Archived from the original on 18 March 2015.

The causative agent and vector of Sleeping Sickness were identified in 1903 by David Bruce, and the subspecies of the protozoa were differentiated in 1910. Bruce had earlier shown that T. brucei was the cause of a similar disease in horses and cattle that was transmitted by the tsetse fly (Glossina morsitans).The first effective treatment, atoxyl, an arsenic-based drug developed by Paul Ehrlich and Kiyoshi Shiga, was introduced in 1910, but blindness was a serious side effect.
- Strong RP (1944). Stitt’s Diagnosis, Prevention and Treatment of Tropical Diseases (Seventh ed.). York, PA: The Blakiston company. p. 165.
Suramin was first synthesized by Oskar Dressel and Richard Kothe in 1916 for Bayer. It was introduced in 1920 to treat the first stage of the disease. By 1922, Suramin was generally combined with tryparsamide (another pentavalent organoarsenic drug), the first drug to enter the nervous system and be useful in the treatment of the second stage of the gambiense form. Tryparsamide was announced in the Journal of Experimental Medicine in 1919 and tested in the Belgian Congo by Louise Pearce of the Rockefeller Institute in 1920. It was used during the grand epidemic in West and Central Africa on millions of people and was the mainstay of therapy until the 1960s. American medical missionary Arthur Lewis Piper was active in using tryparsamide to treat sleeping sickness in the Belgian Congo in 1925. Pentamidine, a highly effective drug for the first stage of the disease, has been used since 1937. During the 1950s, it was widely used as a prophylactic agent in western Africa, leading to a sharp decline in infection rates. At the time, eradication of the disease was thought to be at hand.[citation needed]

German chemist and researcher in the field of azo dyes. He succeeded in chemically producing the active ingredient suramin (germanine), which was used to overcome sleeping sickness .After the Bayer company joined the tar paint industry’s first interest group in 1904, the research team began to work increasingly in the field of drug research. In the middle of the First World War, Oskar Dressel, in collaboration with Richard Kothe and the physician Wilhelm Roehl , succeeded in synthesizing the active ingredient for the drug Bayer 205 in 1916, the first effective cure for tropical trypanosome diseases.
- Steverding D (March 2010). “The development of drugs for treatment of sleeping sickness: a historical review”. Parasites & Vectors. 3 (1): 15. doi:10.1186/1756-3305-3-15. PMC 2848007. PMID 20219092.
- Klingman JD (April 1994). “Arthur Lewis Piper, M.D.: a medical missionary in the Belgian Congo”. Journal of Community Health. 19 (2): 125–46. doi:10.1007/BF02260364. PMID 8006209. S2CID 37502216. Periodicals Archive Online accessed 15 October 2013. Magill AJ, Strickland GT, Maguire JH, Ryan ET, Solomon T (2012). Hunter’s Tropical Medicine and Emerging Infectious Disease (9 ed.). Elsevier Health Sciences. p. 723. ISBN 978-1455740437.
- Steverding D (March 2010). “The development of drugs for treatment of sleeping sickness: a historical review”. Parasites & Vectors. 3 (1): 15. doi:10.1186/1756-3305-3-15. PMC 2848007. PMID 20219092.
Another page says Suramin was first made by the chemists Oskar Dressel, Richard Kothe and Bernhard Heymann at Bayer AG laboratories in Elberfeld, after research on a series of urea-like compounds. The drug is still sold by Bayer under the brand name Germanin. The chemical structure of suramin was kept secret by Bayer for commercial and strategic reasons, but it was elucidated and published in 1924 by Ernest Fourneau and his team at the Pasteur Institute.
- Sneader W (2005). Drug Discovery: A History. John Wiley & Sons. ISBN 9780471899792.
- Fourneau E, Théfouël VJ, Vallée J (1924). “Sur une nouvelle série de médicaments trypanocides”. Comptes Rendus des Séances de l’Académie des Sciences. 178: 675.
Ernest Fourneau (1872 –1949) was a French pharmacist who played a major role in the discovery of synthetic local anesthetics such as amylocaine, as well as in the synthesis of suramin. He authored more than two hundred scholarly works, and has been described as having “helped to establish the fundamental laws of chemotherapy that have saved so many human lives”. Fourneau was a pupil of Friedel and Moureu, and studied in the German laboratories of Ludwig Gattermann in Heidelberg, Hermann Emil Fischer in Berlin and Richard Willstätter in Munich. He headed the research laboratory of Poulenc Frères in Ivry-sur-Seine from 1903 to 1911. One of the products was a synthetic local anesthetic that was named Stovaine (amylocaine). This was a pun on the English translation of “fourneau” as “stove”. (The same pun was used in the brand name of the drug acetarsol, Stovarsol.) Other important medicines were antipyretics. In 1910 Fourneau accepted the directorship of the Pasteur Institute‘s medical chemistry section, with the condition that he maintained his ties with Poulenc Frères. He recruited Germaine Benoit to work in the Institute as a new graduate (methampetamine/sympathomimetic drugs specialist who also worked on sleeping sickness and malaria. In 1947, she was made a Knight of the Légion d’honneur.). Fourneau was a member of the Académie Nationale de Médecine.
- “Fourneau, Ernest”. Complete Dictionary of Scientific Biography, 2008.
- Henry, T. A., “Ernest Fourneau”. 1872–1949. J. Chem. Soc., 1952, pp. 261–272.
- Lesch, John E. (2007), The First Miracle Drugs: How the Sulfa Drugs Transformed Medicine, Oxford University Press, ISBN 978-0-19-518775-5, retrieved 2017-07-02
- Michel, Jean-Marie (2016), “Les établissements Poulenc frères” (PDF), Contribution a l’histoire des polymers en France, Societé Chemique de France, retrieved 2017-07-02
- “Germaine Benoit (1901-1983) – Notice biographique”. webext.pasteur.fr. Retrieved 2022-01-21.
- Lesch, John E. (2007), The First Miracle Drugs: How the Sulfa Drugs Transformed Medicine, Oxford University Press, ISBN 978-0-19-518775-5, retrieved 2017-07-02
- Michel, Jean-Marie (2016), “Les établissements Poulenc frères” (PDF), Contribution a l’histoire des polymers en France, Societé Chemique de France, retrieved 2017-07-02
- Amylocaine was the first synthetic local anesthetic. It was synthesized and patented under the name Stovaine by Ernest Fourneau at the Pasteur Institute in 1903. It was used mostly in spinal anesthesia. Grignard reaction of chloroacetone (1) with one mole of magnesium ethyl bromide gives 1-chloro-2-methyl-butan-2-ol [74283-48-0] (2). Heating with dimethylamine gives 1-(dimethylamino)-2-methylbutan-2-ol [74347-10-7] (3). These two steps can also be treated as interchangeable. Esterification with benzoyl chloride completed the synthesis of amylocaine (4).[ Dimethylaminopivalophenone, an opioid with a similar structure–activity relationship (SAR). It is an amine that is a sole methylene spacer shorter.
- Fourneau, E. (1904). “Stovaïne, anesthésique local”. Bulletin des sciences pharmacologiques. 10: 141-148.
- Debue-Barazer, Christine (2007). “Les Implications scientifiques et industrielles du succès de la Stovaïne : Ernest Fourneau (1872-1949) et la chimie des médicaments en France” Archived 2013-10-05 at the Wayback Machine. Gesnerus 64 (1-2): 24-53.
- Quintard, Jean-Paul; Elissondo, Bernard; Jousseaume, Bernard (1984). “A Convenient Synthesis of N,N-Disubstituted Aminomethyltri-n-butylstannanes, Precursors of the Corresponding Lithium Reagents”. Synthesis. 1984 (6): 495–498. doi:10.1055/s-1984-30879. ISSN 0039-7881. S2CID 95920500.
- Fourneau, Ernest (1904). Comptes rendus hebdomadaires des séances de l’Académie des sciences. Vol. 138. Paris: Academy of Sciences, Centre national de la recherche scientifique (CNRS; French National Centre for Scientific Research). p. 767.
- Zernik, F (1905). “?”. Chem. Zentralbl. 76 (1): 1029.[full citation needed]
- DE169746C, “Patent number DE169746C”. Google Patents.
- DE169787C, “Patent number DE169787C”. Google Patents.
- Smith, Maurice I.; Hatcher, Robert A. (January 1917). “A Contribution to the Pharmacology of Stovaine”. Journal of Pharmacology and Experimental Therapeutics. 9 (4): 231–240.
- Ball, Christine M.; Westhorpe, Rod N. (2004). “Local Anaesthesia after Cocaine”. Anaesthesia and Intensive Care. 32 (2): 157. doi:10.1177/0310057X0403200201. PMID 15957711.
- Dimethylaminopivalophenone is an opioid analgesic with a potency ½ that of morphine.[citation needed] It was initially discovered by Russian scientists in 1954 and subsequently rediscovered in the US in 1969. Its LD50 in mice is 83 mg/kg. It has never been marketed commercially.[citation needed]
- Brewster JH, Eliel EL (2011). “Carbon-Carbon Alkylations with Amines and Ammonium Salts”. Organic Reactions. 7 (3): 99–197. doi:10.1002/0471264180.or007.03. ISBN 9780471264187.
- Kleinman EF (April 2011). “Dimethyl(methylene)ammonium Iodide”. Encyclopedia of Reagents for Organic Synthesis. doi:10.1002/047084289X.rd346. ISBN 978-0471936237.
- Atwal MS, Bauer L, Dixit SN, Gearien JE, Megahy M, Morris R, Pokorny C (November 1969). “Analgetics. II. Relationship between structure and activity of some beta-amino ketones”. Journal of Medicinal Chemistry. 12 (6): 994–7. doi:10.1021/jm00306a006. PMID 5351480.
- “2,2-dimethyl-3-(dimethylamino)-Propiophenone”. ChemIDplus. Retrieved 2 October 2015.
Suramin is also used as a research reagent to inhibit the activation of heterotrimeric G proteins in a variety of GPCRs with varying potency. It prevents the association of heteromeric G proteins and therefore the receptors guanine exchange functionality (GEF). With this blockade the GDP will not release from the Gα subunit so it can not be replaced by a GTP and become activated. This has the effect of blocking downstream G protein mediated signaling of various GPCR proteins including rhodopsin, the A1 adenosine receptor, the D2 receptor, the P2 receptor, and ryanodine receptors.
- Beindl W, Mitterauer T, Hohenegger M, Ijzerman AP, Nanoff C, Freissmuth M (August 1996). “Inhibition of receptor/G protein coupling by suramin analogues”. Molecular Pharmacology. 50 (2): 415–423. PMID 8700151. Archived from the original on 8 September 2017.
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, et al. (September 2006). “International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy”. Pharmacological Reviews. 58 (3): 281–341. doi:10.1124/pr.58.3.3. PMC 3471216. PMID 16968944.
- Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Séguéla P, et al. (March 2001). “International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits”. Pharmacological Reviews. 53 (1): 107–118. PMID 11171941. Archived from the original on 18 November 2016.
- Wolner I, Kassack MU, Ullmann H, Karel A, Hohenegger M (October 2005). “Use-dependent inhibition of the skeletal muscle ryanodine receptor by the suramin analogue NF676”. British Journal of Pharmacology. 146 (4): 525–533. doi:10.1038/sj.bjp.0706359. PMC 1751178. PMID 16056233.
Suramin was studied as a possible treatment for prostate cancer in a clinical trial. Suramin has been studied in a mouse model of autism and in a small phase I/II human trial.
- Ahles TA, Herndon JE, Small EJ, Vogelzang NJ, Kornblith AB, Ratain MJ, et al. (November 2004). “Quality of life impact of three different doses of suramin in patients with metastatic hormone-refractory prostate carcinoma: results of Intergroup O159/Cancer and Leukemia Group B 9480”. Cancer. 101 (10): 2202–2208. doi:10.1002/cncr.20655. PMID 15484217. S2CID 29107328.
- LaFee S, Buschman H (26 May 2017). “Researchers Studying Century-Old Drug in Potential New Approach to Autism”. UC San Diego Health. Archived from the original on 1 June 2017.
- Naviaux JC, Schuchbauer MA, Li K, Wang L, Risbrough VB, Powell SB, Naviaux RK (June 2014). “Reversal of autism-like behaviors and metabolism in adult mice with single-dose antipurinergic therapy”. Translational Psychiatry. 4 (6): e400. doi:10.1038/tp.2014.33. PMC 4080315. PMID 24937094.
- Naviaux RK, Curtis B, Li K, Naviaux JC, Bright AT, Reiner GE, et al. (July 2017). “Low-dose suramin in autism spectrum disorder: a small, phase I/II, randomized clinical trial”. Annals of Clinical and Translational Neurology. 4 (7): 491–505. doi:10.1002/acn3.424. PMC 5497533. PMID 28695149.
- “Q and A – Suramin and Autism | UC San Diego Health”. UC Health – UC San Diego. Retrieved 27 July 2021.
Suramin is a reversible and competitive protein-tyrosine phosphatase (PTPases) inhibitor, also is the potent inhibitor of sirtuins, purified topoisomerase II and SARS-CoV-2 RNA-dependent RNA polymerase (RdRp).[1]
The molecular formula of suramin is C51H40N6O23S6. It is a symmetric molecule in the center of which lies a urea (NH–CO–NH) functional group. Suramin contains six aromatic systems – four benzene rings, sandwiched by a pair of naphthalene moieties – plus four amide functional groups (in addition to the urea) and six sulfonic acid groups. When given as a medication, it is usually delivered as the sodium sulfonate salt as this formulation is water-soluble, though it does deteriorate rapidly in air.
- Phillips MA, Stanley Jr SL (2011). “Chapter 50: Chemotherapy of Protozoal Infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections”. In Brunton LL, Chabner BA, Knollmann BC (eds.). Goodman and Gilman’s The Pharmacological Basis of Therapeutics (12th ed.). McGraw Hill. pp. 1437–1438. ISBN 9780071769396.
The synthesis of suramin itself and structural analogs is by successive formation of the amide bonds from their corresponding amine (aniline) and carboxyl (as acyl chloride) components. Various routes to these compounds have been developed, including starting from separate naphthalene structures and building towards an eventual unification by formation of the urea or starting with a urea and appending successive groups.
- Kassack MU, Braun K, Ganso M, Ullmann H, Nickel P, Böing B, et al. (April 2004). “Structure-activity relationships of analogues of NF449 confirm NF449 as the most potent and selective known P2X1 receptor antagonist”. European Journal of Medicinal Chemistry. 39 (4): 345–357. doi:10.1016/j.ejmech.2004.01.007. PMID 15072843.
- Ullmann H, Meis S, Hongwiset D, Marzian C, Wiese M, Nickel P, et al. (November 2005). “Synthesis and structure-activity relationships of suramin-derived P2Y11 receptor antagonists with nanomolar potency”. Journal of Medicinal Chemistry. 48 (22): 7040–7048. doi:10.1021/jm050301p. PMID 16250663.
- McGeary RP, Bennett AJ, Tran QB, Prins J, Ross BP (2009). “An ‘inside-out’ approach to suramin analogues”. Tetrahedron. 65 (20): 3990–3997. doi:10.1016/j.tet.2009.03.033.
Another drug, the organoarsenical melarsoprol (Arsobal) developed in the 1940s is effective for people with second-stage sleepingsickness. However, 3–10% of those injected have reactive encephalopathy (convulsions, progressive coma, or psychotic reactions), and 10–70% of such cases result in death; it can cause brain damage in those who survive the encephalopathy. However, due to its effectiveness, melarsoprol is still used today. Resistance to melarsoprol is increasing, and combination therapy with nifurtimox is currently under research.[citation needed]
Melarsoprol has a high number of side effects. Common side effects include brain dysfunction, numbness, rashes, and kidney and liver problems. About 1-5% of people die during treatment, although this is tolerated due to sleeping sickness itself having a practically 100% mortality rate when untreated. In those with glucose-6-phosphate dehydrogenase (G6PD) deficiency, red blood cell breakdown may occur. It has not been studied in pregnancy. It works by blocking pyruvate kinase, an enzyme required for aerobic metabolism by the parasite. Melarsoprol has been used medically since 1949. It is on the World Health Organization’s List of Essential Medicines. In regions of the world where the disease is common, melarsoprol is provided for free by the World Health Organization. It is not commercially available in Canada or the United States. In the United States, it may be obtained from the Centers for Disease Control and Prevention, while in Canada it is available from Health Canada.
- “Our Formulary Infectious Diseases Laboratories CDC”. www.cdc.gov. 22 September 2016. Archived from the original on 16 December 2016. Retrieved 7 December 2016.
- “Melarsoprol Drug Information, Professional”. www.drugs.com. 20 December 1994. Archived from the original on 30 December 2016. Retrieved 7 December 2016.
- “WHO Model Prescribing Information: Drugs Used in Parasitic Diseases – Second Edition: Protozoa: African trypanosomiasis: Melarsoprol”. WHO. 1995. Archived from the original on 2016-11-10. Retrieved 2016-11-09.
- “Trypanosomiasis, human African (sleeping sickness)”. World Health Organization. February 2016. Archived from the original on 4 December 2016. Retrieved 7 December 2016.
Melarsoprol is a prodrug, a complex of melarsen oxide (a melamine derivative of phenylarsonous acid) with dimercaprol (also known as British anti-Lewisite, or BAL). It is metabolized to melarsen oxide in the body, which then acts by irreversibly binding to sulfhydryl groups on the enzyme pyruvate kinase, thus disrupting energy production in the parasite. The inability to distinguish between the host’s and the parasite’s pyruvate kinase renders this drug highly toxic, with many side effects.[citation needed]
Melarsen oxide also reacts with trypanothione (a spermidine-glutathione adduct that replaces glutathione in trypanosomes). It forms a melarsen oxide-trypanothione adduct (Mel T) that competitively inhibits trypanothione reductase, effectively killing the protist.
- Brunton L (2011). Goodman & Gillman’s The Pharmacological Basis of Therapeutics. McGraw Hill Medical. pp. 1427–28.
- Trypanothione is an unusual form of glutathione containing two molecules of glutathione joined by a spermidine (polyamine) linker. It is found in parasitic protozoa such as leishmania and trypanosomes. These protozoal parasites are the cause of leishmaniasis, sleeping sickness and Chagas’ disease. Trypanothione was discovered by Alan Fairlamb. Its structure was proven by chemical synthesis. It is present mainly in the Kinetoplastida but can be found in other parasitic protozoa such as Entamoeba histolytica. Since this thiol is absent from humans and is essential for the survival of the parasites, the enzymes that make and use this molecule are targets for the development of new drugs to treat these diseases.
- Fairlamb AH, Cerami A (1992). “Metabolism and functions of trypanothione in the Kinetoplastida”. Annu. Rev. Microbiol. 46: 695–729. doi:10.1146/annurev.mi.46.100192.003403. PMID 1444271.
- Fairlamb, A. H.; Blackburn, P.; Ulrich, P.; Chait, B. T.; Cerami, A. (Mar 1985). “Trypanothione: a novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids”. Science. 227 (4693): 1485–1487. Bibcode:1985Sci…227.1485F. doi:10.1126/science.3883489. ISSN 0036-8075. PMID 3883489.
- Ondarza, Raul (2005). “Identification of trypanothione from the human pathogen Entamoeba histolytica by mass spectrometry and chemical analysis”. Biotechnol. Appl. Biochem. 42 (Pt 2): 175–181. doi:10.1042/BA20050023. PMID 15801913. S2CID 23482542.
- Schmidt A, Krauth-Siegel RL (November 2002). “Enzymes of the trypanothione metabolism as targets for antitrypanosomal drug development”. Curr Top Med Chem. 2 (11): 1239–59. doi:10.2174/1568026023393048. PMID 12171583.
- Trypanothione-dependent enzymes include reductases, peroxidases, glyoxalases and transferases. Trypanothione-disulfide reductase (TryR) was the first trypanothione-dependent enzyme to be discovered (EC 1.8.1.12). It is an NADPH-dependent flavoenzyme that reduces trypanothione disulfide. TryR is essential for survival of these parasites both in vitro and in the human host.
- Tovar J, Wilkinson S, Mottram JC, Fairlamb AH (July 1998). “Evidence that trypanothione reductase is an essential enzyme in Leishmania by targeted replacement of the tryA gene locus”. Mol. Microbiol. 29 (2): 653–60. doi:10.1046/j.1365-2958.1998.00968.x. PMID 9720880.
- Krieger S, Schwarz W, Ariyanayagam MR, Fairlamb AH, Krauth-Siegel RL, Clayton C (February 2000). “Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress”. Mol. Microbiol. 35 (3): 542–52. doi:10.1046/j.1365-2958.2000.01721.x. PMID 10672177.
- A major function of trypanothione is in the defence against oxidative stress. Here, trypanothione-dependent enzymes such as tryparedoxin peroxidase (TryP) reduce peroxides using electrons donated either directly from trypanothione, or via the redox intermediate tryparedoxin (TryX). Trypanothione-dependent hydrogen peroxide metabolism is particularly important in these organisms because they lack catalase. Since the trypanosomatids also lack an equivalent of thioredoxin reductase, trypanothione reductase is the sole path that electrons can take from NADPH to these antioxidant enzymes.
Drug resistance evolution is encouraged by the use of diminazene for nagana. Animal trypanosomiasis, also known as nagana and nagana pest, or sleeping sickness, is a disease of vertebrates. The disease is caused by trypanosomes of several species in the genus Trypanosoma such as T. brucei. T. vivax causes nagana mainly in West Africa, although it has spread to South America. The trypanosomes infect the blood of the vertebrate host, causing fever, weakness, and lethargy, which lead to weight loss and anemia; in some animals the disease is fatal unless treated. When an infected tsetse fly bites an animal, the parasites are transmitted through its saliva. It can also be spread by fomites such as surgical instruments, needles, and syringes.
- Maudlin I (December 2006). “African trypanosomiasis”. Annals of Tropical Medicine and Parasitology. Taylor & Francis. 100 (8): 679–701. doi:10.1179/136485906×112211. PMID17227648. S2CID19567898. …cites this study: Fèvre EM, Coleman PG, Odiit M, Magona JW, Welburn SC, Woolhouse ME (August 2001). “The origins of a new Trypanosoma brucei rhodesiense sleeping sickness outbreak in eastern Uganda”. Lancet. Elsevier BV. 358 (9282): 625–628. doi:10.1016/s0140-6736(01)05778-6. PMID11530149. S2CID205937568.
- Batista, Jael S; Rodrigues, Carla MF; García, Herakles A; Bezerra, Francisco SB; Olinda, Robério G; Teixeira, Marta MG; Soto-Blanco, Benito (May 2011). “Association of Trypanosoma vivax in extracellular sites with central nervous system lesions and changes in cerebrospinal fluid in experimentally infected goats”. Veterinary Research. 42 (1): 63. doi:10.1186/1297-9716-42-63. PMC 3105954. PMID 21569364.
Eflornithine (difluoromethylornithine or DFMO), the most modern treatment, was developed in the 1970s by Albert Sjoerdsma and underwent clinical trials in the 1980s. The drug was approved by the United States Food and Drug Administration in 1990. Aventis, the company responsible forits manufacture, halted production in 1999. In 2001, Aventis, in association with Médecins Sans Frontières and the World Health Organization, signed a long-term agreement to manufacture and donate the drug.[citation needed]In addition to sleeping sickness, previous names have included negro lethargy, maladie du sommeil (Fr), Schlafkrankheit (Ger), African lethargy, and Congo trypanosomiasis.
Eflornithine was initially developed for cancer treatment at Merrell Dow Research Institute in the late 1970s, but was found to be ineffective in treating malignancies. However, it was discovered to be highly effective in reducing hair growth, as well as in the treatment of African trypanosomiasis (sleeping sickness), especially the West African form (Trypanosoma brucei gambiense).
- Wolf JE, Shander D, Huber F, Jackson J, Lin CS, Mathes BM, Schrode K (January 2007). “Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair”. International Journal of Dermatology. 46 (1): 94–98. doi:10.1111/j.1365-4632.2006.03079.x. PMID 17214730. S2CID 10795478.
- Pepin J, Milord F, Guern C, Schechter PJ (December 1987). “Difluoromethylornithine for arseno-resistant Trypanosoma brucei gambiense sleeping sickness”. Lancet. 2 (8573): 1431–1433. doi:10.1016/S0140-6736(87)91131-7. PMID 2891995. S2CID 41019313.
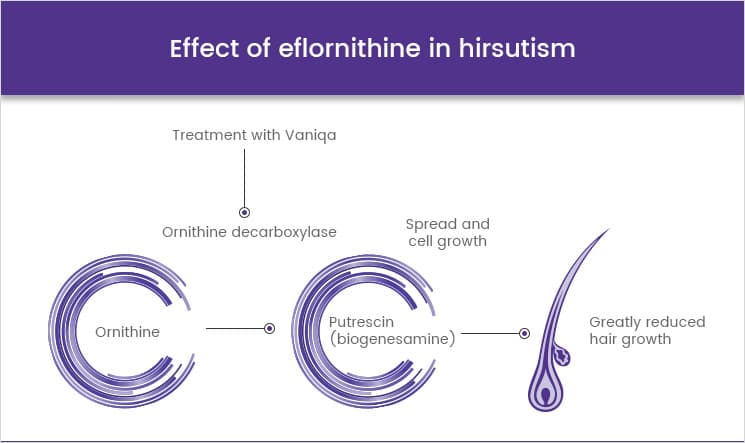
In the 1980s, Gillette was awarded a patent for the discovery that topical application of eflornithine HCl cream inhibits hair growth. In the 1990s, Gillette conducted dose-ranging studies with eflornithine in hirsute women that demonstrated that the drug slows the rate of facial hair growth. Gillette then filed a patent for the formulation of eflornithine cream. In July 2000, the U.S. Food and Drug Administration (FDA) granted a New Drug Application for Vaniqa. The following year, the European Commission issued its Marketing Authorisation.[citation needed]
- Hellgren U, Ericsson O, AdenAbdi Y, Gustafsson LL (20 May 2003). Handbook of Drugs for Tropical Parasitic Infections. CRC Press. p. 60. ISBN 9780203211519.
- Robinson, Victor, ed. (1939). “African Lethargy, Sleeping Sickness, or Congo trypanosomiasis; Trypanosoma gambiense”. The Modern Home Physician, A New Encyclopedia of Medical Knowledge. WM. H. Wise & Company (New York)., pp. 20–21.
- Strong RP (1944). Stitt’s Diagnosis, Prevention and Treatment of Tropical Diseases (Seventh ed.). York, PA: The Blakiston company. p. 164.
Tryptophol forms in the liver as a side-effect of disulfiram treatment.
- Cornford, E. M.; Bocash, W. D.; Braun, L. D.; Crane, P. D.; Oldendorf, W. H.; MacInnis, A. J. (1979). “Rapid distribution of tryptophol (3-indole ethanol) to the brain and other tissues”. Journal of Clinical Investigation. 63 (6): 1241–1248. doi:10.1172/JCI109419. PMC 372073. PMID 447842.
Disulfiram is a medication used to support the treatment of chronic alcoholism by producing an acute sensitivity to ethanol (drinking alcohol). Disulfiram works by inhibiting the enzymealdehyde dehydrogenase, causing many of the effects of a hangover to be felt immediately following alcohol consumption. Disulfiram plus alcohol, even small amounts, produces flushing, throbbing in the head and neck, a throbbing headache, respiratory difficulty, nausea, copious vomiting, sweating, thirst, chest pain, palpitation, dyspnea, hyperventilation, fast heart rate, low blood pressure, fainting, marked uneasiness, weakness, vertigo, blurred vision, and confusion. In severe reactions there may be respiratory depression, cardiovascular collapse, abnormal heart rhythms, heart attack, acute congestive heart failure, unconsciousness, convulsions, and death.
- “Antabuse – disulifram tablet”. DailyMed. National Institutes of Health. May 23, 2016. Retrieved 4 July 2016.
In the body, alcohol is converted to acetaldehyde, which is then broken down by acetaldehyde dehydrogenase. When the dehydrogenase enzyme is inhibited, acetaldehyde builds up, causing unpleasant side effects (Disulfiram-alcohol reaction).
Once disulfiram-treated patients take alcohol, even in small doses, they experience strong unpleasant sensations (flush, nausea, lightheadedness, headache, sweating, vomiting, and vertigo).
- Wright C, Moore RD (June 1990). “Disulfiram treatment of alcoholism”. The American Journal of Medicine. 88 (6): 647–655. doi:10.1016/0002-9343(90)90534-K. PMID 2189310.
Disulfiram has been used to treat alcoholism since 1948 after its accidental discovery in Denmark. Disulfiram is used as a second-line treatment, behind acamprosate and naltrexone, for alcohol dependence.
- Murthy, K. Krishna; Praveenlal, K. (July 1988). “An Experience with Disulfiram in the management of Alcohol Dependence Syndrome”. Indian Journal of Psychological Medicine. 11 (2): 145–148. doi:10.1177/0975156419880213. S2CID 220676012.
- Altun, Gurcan; Altun, Armagan; Erdogan, Okan (October 2006). “Acute Myocardial Infarction Due to Disulfiram (Antabus)–Alcohol Interaction”. Cardiovascular Drugs and Therapy. 20 (5): 391–392. doi:10.1007/s10557-006-0493-8. PMID 17119876. S2CID 30117045. ProQuest 213848944.
- Glatt, M. M. (April 1959). “Disulfiram and Citrated Calcium Carbimide in the Treatment of Alcoholism”. Journal of Mental Science. 105 (439): 476–481. doi:10.1192/bjp.105.439.476. PMID 13665310.
- Stokes M, Abdijadid S (January 2018). “Disulfiram”. Stat Pearls. Treasure Island (FL): StatPearls Publishing. PMID29083801
Under normal metabolism, alcohol is broken down in the liver by the enzyme alcohol dehydrogenase to acetaldehyde, which is then converted by the enzyme acetaldehyde dehydrogenase to a harmless acetic acid derivative (acetyl coenzyme A). Disulfiram blocks this reaction at the intermediate stage by blocking acetaldehyde dehydrogenase. After alcohol intake under the influence of disulfiram, the concentration of acetaldehyde in the blood may be five to 10 times higher than that found during metabolism of the same amount of alcohol alone. As acetaldehyde is one of the major causes of the symptoms of a “hangover“, this produces immediate and severe negative reaction to alcohol intake. About 5 to 10 minutes after alcohol intake, the patient may experience the effects of a severe hangover for a period of 30 minutes up to several hours. Symptoms usually include flushing of the skin, accelerated heart rate, low blood pressure, nausea, and vomiting. Uncommon adverse events include shortness of breath, throbbing headache, visual disturbance, mental confusion, postural syncope, and circulatory collapse.
- Yourick JJ, Faiman MD (November 1987). “Diethyldithiocarbamic acid-methyl ester: a metabolite of disulfiram and its alcohol sensitizing properties in the disulfiram-ethanol reaction”. Alcohol. 4 (6): 463–467. doi:10.1016/0741-8329(87)90086-3. PMID 2829942.
Disulfiram should not be taken if alcohol has been consumed in the last 12 hours. There is no tolerance to disulfiram: the longer it is taken, the stronger its effects. As disulfiram is absorbed slowly through the digestive tract and eliminated slowly by the body, the effects may last for up to two weeks after the initial intake; consequently, medical ethics dictate that patients must be fully informed about the disulfiram-alcohol reaction.
- “Disulfiram Official FDA information, side effects and uses”. Drugs.com. Retrieved 2011-04-11.
- Wright C, Moore RD (June 1990). “Disulfiram treatment of alcoholism”. The American Journal of Medicine. 88 (6): 647–655. doi:10.1016/0002-9343(90)90534-K. PMID 2189310.
- “Antabuse – disulifram tablet”. DailyMed. National Institutes of Health. May 23, 2016. Retrieved 4 July 2016.
Disulfiram does not reduce alcohol cravings, so a major problem associated with this drug is extremely poor compliance. Methods to improve compliance include subdermal implants, which release the drug continuously over a period of up to 12 weeks, and supervised administration practices, for example, having the drug regularly administered by one’s spouse.[medical citation needed]
Although disulfiram remained the most common pharmaceutical treatment of alcohol abuse until the end of the 20th century, today it is often replaced or accompanied with newer drugs, primarily the combination of naltrexone and acamprosate, which directly attempt to address physiological processes in the brain associated with alcohol abuse.[citation needed]
Side effects
The most common side effects in the absence of alcohol are headache, and a metallic or garlic taste in the mouth, though more severe side effects may occur. Tryptophol, a chemical compound that induces sleep in humans, is formed in the liver after disulfiram treatment. Less common side effects include decrease in libido, liver problems, skin rash, and nerve inflammation. Liver toxicity is an uncommon but potentially serious side effect, and risk groups e.g. those with already impaired liver function should be monitored closely. That said, the rate of disulfiram-induced hepatitis are estimated to be in between 1 per 25,000 to 1 in 30,000, and rarely the primary cause for treatment cessation.
- “Disulfiram Side Effects”. Drugs.com. Retrieved 6 November 2010.
- Cornford EM, Bocash WD, Braun LD, Crane PD, Oldendorf WH, MacInnis AJ (June 1979). “Rapid distribution of tryptophol (3-indole ethanol) to the brain and other tissues”. The Journal of Clinical Investigation. 63 (6): 1241–1248. doi:10.1172/JCI109419. PMC 372073. PMID 447842.
- “Antabuse (disulfiram)”. netdoctor. November 18, 2013. Retrieved April 28, 2017.
- Center for Substance Abuse Treatment (2009). Chapter 3—Disulfiram. Substance Abuse and Mental Health Services Administration (US).
Cases of disulfiram neurotoxicity have also occurred, causing extrapyramidal and other symptoms. Disulfiram can produce neuropathy in daily doses of less than the usually recommended 500 mg. Nerve biopsies showed axonal degeneration and the neuropathy is difficult to distinguish from that associated with ethanol abuse. Disulfiram neuropathy occurs after a variable latent period (mean 5 to 6 months) and progresses steadily. Slow improvement may occur when the drug’s use is stopped; often there is complete recovery eventually.
- Boukriche Y, Weisser I, Aubert P, Masson C (September 2000). “MRI findings in a case of late onset disulfiram-induced neurotoxicity”. Journal of Neurology. 247 (9): 714–715. doi:10.1007/s004150070119. PMID 11081815. S2CID 1982036.
- Watson CP, Ashby P, Bilbao JM (July 1980). “Disulfiram neuropathy”. Canadian Medical Association Journal. 123 (2): 123–126. PMC 1704662. PMID 6266628.
Disulfiram disrupts metabolism of several other compounds, including paracetamol (acetaminophen), theophylline and caffeine. However, in most cases, this disruption is mild and presents itself as a 20–40% increase in the half-life of the compound at typical dosages of disulfiram.[citation needed]
- Poulsen HE, Ranek L, Jørgensen L (February 1991). “The influence of disulfiram on acetaminophen metabolism in man”. Xenobiotica; the Fate of Foreign Compounds in Biological Systems. 21 (2): 243–249. doi:10.3109/00498259109039466. PMID 2058179.
- Loi CM, Day JD, Jue SG, Bush ED, Costello P, Dewey LV, Vestal RE (May 1989). “Dose-dependent inhibition of theophylline metabolism by disulfiram in recovering alcoholics”. Clinical Pharmacology and Therapeutics. 45 (5): 476–486. doi:10.1038/clpt.1989.61. PMID 2721103. S2CID 39324339.
- Beach CA, Mays DC, Guiler RC, Jacober CH, Gerber N (March 1986). “Inhibition of elimination of caffeine by disulfiram in normal subjects and recovering alcoholics”. Clinical Pharmacology and Therapeutics. 39 (3): 265–270. doi:10.1038/clpt.1986.37. PMID 3948467. S2CID 29110467.
- Theophylline, also known as 1,3-dimethylxanthine, is a drug that inhibits phosphodiesterase and blocks adenosine receptors. It is used to treat chronic obstructive pulmonary disease (COPD) and asthma. Its pharmacology is similar to other methylxanthine drugs (e.g., theobromine and caffeine). Trace amounts of theophylline are naturally present in tea, coffee, chocolate, yerba maté, guarana, and kola nut.
- “Theophylline”. PubChem, US National Library of Medicine. 26 August 2023. Retrieved 2 September 2023.
- Barnes PJ (October 2013). “Theophylline”. American Journal of Respiratory and Critical Care Medicine. 188 (8): 901–906. doi:10.1164/rccm.201302-0388PP. PMID 23672674.
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans (1991). “Coffee, Tea, Mate, Methylxanthines and Methylglyoxal”. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. International Agency for Research on Cancer. 51: 391–419. PMC 7681294. PMID 2033791.
‘Disulfiram effect’ and similarly acting substances
In medicine, the term “disulfiram effect” refers to an adverse effect of a particular medication in causing an unpleasant hypersensitivity to alcohol, similar to the effect caused by disulfiram administration.[citation needed]
Examples:
- Antibiotics (nitroimidazoles), e.g. metronidazole
- First-generation sulfonylureas, e.g. tolbutamide and chlorpropamide
- Several cephalosporin drugs, including cefoperazone, cefamandole and cefotetan, that have a N-methylthio-tetrazole moiety
- Griseofulvin, an oral antifungal drug
- Procarbazine
- Temposil, or citrated calcium carbimide, has the same function as disulfiram, but is weaker and safer.[citation needed]
- Coprine, which metabolizes to 1-aminocyclopropanol, a chemical having the same metabolic effects as disulfiram. It occurs naturally in the otherwise edible common ink cap mushroom (Coprinopsis atramentaria), hence its colloquial name “tippler’s bane”. Similar reactions have been recorded with Clitocybe clavipes and Suillellus luridus, although the agent in those species is unknown.
History of disulfiram
The synthesis of disulfiram, originally known as tetraethylthiuram disulfide, was first reported in 1881. By around 1900, it was introduced to the industrial process of sulfur vulcanization of rubber and became widely used. In 1937 a plant physician in the American rubber industry described adverse reactions to alcohol in workers exposed to tetramethylthiuram monosulfide and disulfide, and proposed that this effect of disulfiram and related compounds might lead to ”the cure for alcoholism”; the effect was also noticed in workers at a Swedish rubber boot factory.
- Kragh H (2008). “From Disulfiram to Antabuse: The Invention of a Drug” (PDF). Bulletin for the History of Chemistry. 33 (2): 82–88.
In the early 1940s it had been tested as a treatment for scabies, a parasitic skin infection, as well as intestinal worms.
- Kragh H (2008). “From Disulfiram to Antabuse: The Invention of a Drug” (PDF). Bulletin for the History of Chemistry. 33 (2): 82–88.
Around that time, during the German occupation of Denmark, Erik Jacobsen and Jens Hald at the Danish drug company Medicinalco picked up on that research and began exploring the use of disulfiram to treat intestinal parasites. The company had a group of enthusiastic self-experimenters that called itself the “Death Battalion”, and in the course of testing the drug on themselves, accidentally discovered that drinking alcohol while the drug was still in their bodies made them mildly sick.
- Kragh H (2008). “From Disulfiram to Antabuse: The Invention of a Drug” (PDF). Bulletin for the History of Chemistry. 33 (2): 82–88.
- Altman LK (1998). Who Goes First?: The Story of Self-Experimentation in Medicine. University of California Press. ISBN 9780520212817.
They made that discovery in 1945, and did nothing with it until two years later, when Jacobsen gave an impromptu talk and mentioned that work, which was discussed afterwards in newspapers at the time, leading them to further explore the use of the drug for that purpose. That work included small clinical trials with Oluf Martensen-Larsen, a doctor who worked with alcoholics. They published their work starting in 1948.
- Kragh H (2008). “From Disulfiram to Antabuse: The Invention of a Drug” (PDF). Bulletin for the History of Chemistry. 33 (2): 82–88.
- Altman LK (1998). Who Goes First?: The Story of Self-Experimentation in Medicine. University of California Press. ISBN 9780520212817.
- Hald J, Jacobsen E, Larsen V (July 1948). “The Sensitizing Effect of Tetraethylthiuramdisulphide (Antabuse) to Ethylalcohol”. Acta Pharmacologica et Toxicologica. 4 (3–4): 285–296. doi:10.1111/j.1600-0773.1948.tb03350.x.
The chemists at Medicinalco discovered a new form of disulfiram while trying to purify a batch that had been contaminated with copper. This form turned out to have better pharmacological properties, and the company patented it and used that form for the product that was introduced as Antabus (later anglicized to Antabuse). This work led to renewed study of the human metabolism of ethanol. It was already known that ethanol was mostly metabolized in the liver, with it being converted first to acetaldehyde and then acetaldehyde to acetic acid and carbon dioxide, but the enzymes involved were not known. By 1950 the work led to the knowledge that ethanol is oxidized to acetaldehyde by alcohol dehydrogenase and acetaldehyde is oxidized to acetic acid by aldehyde dehydrogenase (ALDH), and that disulfiram works by inhibiting ALDH, leading to a buildup of acetaldehyde, which is what causes the negative effects in the body.
- Kragh H (2008). “From Disulfiram to Antabuse: The Invention of a Drug” (PDF). Bulletin for the History of Chemistry. 33 (2): 82–88.
The drug was first marketed in Denmark and as of 2008, Denmark is the country where it is most widely prescribed. It was approved by the FDA in 1951. The FDA later approved other drugs for treatment of alcoholism, namely naltrexone in 1994 and acamprosate in 2004.
- Kragh H (2008). “From Disulfiram to Antabuse: The Invention of a Drug” (PDF). Bulletin for the History of Chemistry. 33 (2): 82–88.
- “New Drug Application (NDA) 007883: Disulfiram (Antabuse)”. FDA-Approved Drugs. U.S. Food and Drug Administration (FDA). Retrieved 4 August 2018.
Though the Occupational Safety and Health Administration (OSHA) in the US has not set a permissible exposure limit (PEL) for disulfiram in the workplace, the National Institute for Occupational Safety and Health has set a recommended exposure limit (REL) of 2 mg/m3 and recommended that workers avoid concurrent exposure to ethylene dibromide.
- NIOSH Pocket Guide to Chemical Hazards. “#0244”. National Institute for Occupational Safety and Health (NIOSH).
Disulfiram Research
Disulfiram has been studied as a possible treatment for cancer, parasitic infections, anxiety disorder, and latent HIV infection.
- Jiao Y, Hannafon BN, Ding WQ (2016). “Disulfiram’s Anticancer Activity: Evidence and Mechanisms”. Anti-Cancer Agents in Medicinal Chemistry. 16 (11): 1378–1384. doi:10.2174/1871520615666160504095040. PMID 27141876.
- Shirley DA, Sharma I, Warren CA, Moonah S (2021). “Drug Repurposing of the Alcohol Abuse Medication Disulfiram as an Anti-Parasitic Agent”. Frontiers in Cellular and Infection Microbiology. 11: 633194. doi:10.3389/fcimb.2021.633194. PMC 7991622. PMID 33777846.
- Gallagher A (23 April 2022). “Drug Used to Treat Alcoholism Shows Promise for Anxiety, Study Results Show”. Pharmacy Times. Retrieved 2022-04-25.
- Rasmussen TA, Lewin SR (July 2016). “Shocking HIV out of hiding: where are we with clinical trials of latency reversing agents?”. Current Opinion in HIV and AIDS. 11 (4): 394–401. doi:10.1097/COH.0000000000000279. PMID 26974532. S2CID 25995091.
Disulfiram has shown reversing of retinitis pigmentosa in rats.
- “A key to restoring sight may be held in a drug that treats alcoholism”. URMC Newsroom. Retrieved 2022-04-13.
- Telias M, Sit KK, Frozenfar D, Smith B, Misra A, Goard MJ, Kramer RH (March 2022). “Retinoic acid inhibitors mitigate vision loss in a mouse model of retinal degeneration”. Science Advances. 8 (11): eabm4643. Bibcode:2022SciA….8M4643T. doi:10.1126/sciadv.abm4643. PMC 8932665. PMID 35302843.
- Papadopoulos L (2022-03-19). “A drug once used to treat alcoholism may cure retinal degeneration”. interestingengineering.com. Retrieved 2022-04-13.
Cancer
When disulfiram creates complexes with metals (dithiocarbamate complexes), it is a proteasome inhibitor and as of 2016 it had been studied in in vitro experiments, model animals, and small clinical trials as a possible treatment for liver metastasis, metastatic melanoma, glioblastoma, non-small cell lung cancer, and prostate cancer. Various clinical trials of copper depletion agents have been carried out.[citation needed]
- Cvek B, Dvorak Z (August 2008). “The value of proteasome inhibition in cancer. Can the old drug, disulfiram, have a bright new future as a novel proteasome inhibitor?”. Drug Discovery Today. 13 (15–16): 716–722. doi:10.1016/j.drudis.2008.05.003. PMID 18579431.
- Jiao Y, Hannafon BN, Ding WQ (2016). “Disulfiram’s Anticancer Activity: Evidence and Mechanisms”. Anti-Cancer Agents in Medicinal Chemistry. 16 (11): 1378–1384. doi:10.2174/1871520615666160504095040. PMID 27141876.
Parasitic infections
In the body, disulfiram is rapidly metabolized to diethyldithiocarbamate (ditiocarb), which binds to metal ions such as zinc or copper to form zinc or copper diethyldithiocarbamate (zinc or copper ditiocarb). The zinc diethyldithiocarbamate (zinc-ditiocarb) metabolite of disulfiram is extremely potent against the diarrhea and liver abscess-causing parasite Entamoeba histolytica and might be active against other deadly parasites.
- Shirley DA, Sharma I, Warren CA, Moonah S (2021). “Drug Repurposing of the Alcohol Abuse Medication Disulfiram as an Anti-Parasitic Agent”. Frontiers in Cellular and Infection Microbiology. 11: 633194. doi:10.3389/fcimb.2021.633194. PMC 7991622. PMID 33777846.
- Ghosh S, Farr L, Singh A, Leaton LA, Padalia J, Shirley DA, et al. (September 2020). “COP9 signalosome is an essential and druggable parasite target that regulates protein degradation”. PLOS Pathogens. 16 (9): e1008952. doi:10.1371/journal.ppat.1008952. PMC 7531848. PMID 32960936.
HIV
Disulfiram has also been identified by systematic high-throughput screening as a potential HIV latency reversing agent (LRA). Reactivation of latent HIV infection in patients is part of an investigational strategy known as “shock and kill” which may be able to reduce or eliminate the HIV reservoir. Recent phase II dose-escalation studies in patients with HIV who are controlled on antiretroviral therapy have observed an increase in cell-associated unspliced HIV RNA with increasing exposure to disulfiram and its metabolites. Disulfiram is also being investigated in combination with vorinostat, another investigational latency reversing agent, to treat HIV.
- Lee SA, Elliott JH, McMahon J, Hartogenesis W, Bumpus NN, Lifson JD, et al. (March 2019). “Population Pharmacokinetics and Pharmacodynamics of Disulfiram on Inducing Latent HIV-1 Transcription in a Phase IIb Trial”. Clinical Pharmacology and Therapeutics. 105 (3): 692–702. doi:10.1002/cpt.1220. PMC 6379104. PMID 30137649.
- Xing S, Bullen CK, Shroff NS, Shan L, Yang HC, Manucci JL, et al. (June 2011). “Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation”. Journal of Virology. 85 (12): 6060–6064. doi:10.1128/JVI.02033-10. PMC 3126325. PMID 21471244.
- Knights HD (2017). “A Critical Review of the Evidence Concerning the HIV Latency Reversing Effect of Disulfiram, the Possible Explanations for Its Inability to Reduce the Size of the Latent Reservoir In Vivo, and the Caveats Associated with Its Use in Practice”. AIDS Research and Treatment. 2017: 8239428. doi:10.1155/2017/8239428. PMC 5390639. PMID 28465838.
- Clinical trial number NCT03198559 for “Combination Latency Reversal With High Dose Disulfiram Plus Vorinostat in HIV-infected Individuals on ART” at ClinicalTrials.gov
- Rasmussen TA, Lewin SR (July 2016). “Shocking HIV out of hiding: where are we with clinical trials of latency reversing agents?”. Current Opinion in HIV and AIDS. 11 (4): 394–401. doi:10.1097/COH.0000000000000279. PMID 26974532. S2CID 25995091.
COVID-19
Disulfiram has been shown to inhibit the papain-like proteases of MERS-CoV and SARS-CoV. It has been examined in a small inconclusive retrospective observational study for its effects on COVID-19 symptoms. In a small randomized controlled trial that has yet to be published in a medical journal, seven COVID patients received disulfiram. No serious adverse events were reported in these patients.
- Lin MH, Moses DC, Hsieh CH, Cheng SC, Chen YH, Sun CY, Chou CY (February 2018). “Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes”. Antiviral Research. 150: 155–163. doi:10.1016/j.antiviral.2017.12.015. PMC 7113793. PMID 29289665.
- Tamburin S, Mantovani E, De Bernardis E, Zipeto D, Lugoboni F, et al. (Gruppo InterSERT di Collaborazione Scientifica (GICS)) (September 2021). “COVID-19 and related symptoms in patients under disulfiram for alcohol use disorder”. Internal and Emergency Medicine. 16 (6): 1729–1731. doi:10.1007/s11739-021-02633-y. PMC 7814518. PMID 33464469.
- Clinical trial number NCT04485130 for “DISulfiram for COvid-19 (DISCO) Trial” at ClinicalTrials.gov
Natural occurrences of Tryptophol
Tryptophol can be found in Pinus sylvestris needles or seeds. It is produced by the trypanosomal parasite (Trypanosoma brucei) in sleeping sickness (African trypanosomiasis). Tryptophol is found in wine and beer as a secondary product of ethanol fermentation (a product also known as congener) by Saccharomyces cerevisiae. It is also an autoantibiotic produced by the fungus Candida albicans. It can also be isolated from the marine sponge Ircinia spiculosa.
- Sandberg, Göran (1984). “Biosynthesis and metabolism of indole-3-ethanol and indole-3-acetic acid by Pinus sylvestris L. Needles”. Planta. 161 (5): 398–403. doi:10.1007/BF00394569. PMID 24253838. S2CID 23500542.
- Sandberg, Goran; Ernstsen, Arild; Hamnede, Marianne (1987). “Dynamics of indole-3-acetic acid and indole-3-ethanol during development and germination of Pinus sylvestris seeds”. Physiologia Plantarum. 71 (4): 411–418. doi:10.1111/j.1399-3054.1987.tb02876.x.
- Richard Seed, John; Seed, Thomas M.; Sechelski, John (1978). “The biological effects of tryptophol (indole-3-ethanol): Hemolytic, biochemical and behavior modifying activity”. Comparative Biochemistry and Physiology C. 60 (2): 175–185. doi:10.1016/0306-4492(78)90091-6. PMID 28889.
- Gil, C.; Gómez-Cordovés, C. (1986). “Tryptophol content of young wines made from Tempranillo, Garnacha, Viura and Airén grapes”. Food Chemistry. 22: 59–65. doi:10.1016/0308-8146(86)90009-9.
- Ribéreau-Gayon, P; Sapis, JC (1965). “On the presence in wine of tyrosol, tryptophol, phenylethyl alcohol and gamma-butyrolactone, secondary products of alcoholic fermentation”. Comptes Rendus de l’Académie des Sciences, Série D. 261 (8): 1915–6. PMID 4954284. (Article in French)
- Lingappa, BT; Prasad, M; Lingappa, Y; Hunt, DF; Biemann, K (1969). “Phenethyl alcohol and tryptophol: Autoantibiotics produced by the fungus Candida albicans”. Science. 163 (3863): 192–4. Bibcode:1969Sci…163..192L. doi:10.1126/science.163.3863.192. PMID 5762768. S2CID 12430791.
- Erdoğan, İlkay; Sener, B; Higa, T (2000). “Tryptophol, a plant auxin isolated from the marine sponge Ircinia spinulosa”. Biochemical Systematics and Ecology. 28 (8): 793–794. doi:10.1016/S0305-1978(99)00111-8. PMID 10856636.
Metabolism of Tryptophol
Biosynthesis
It was first described by Felix Ehrlich in 1912 (the same Ehrlich who discovered the amino acid isoleucine in hemoglobin in 1903). Ehrlich demonstrated that yeast attacks the natural amino acids essentially by splitting off carbon dioxide and replacing the amino group with hydroxyl. By this reaction, tryptophan gives rise to tryptophol. Tryptophan is first deaminated to 3-indolepyruvate. It is then decarboxylated to indole acetaldehyde by indolepyruvate decarboxylase. This latter compound is transformed to tryptophol by alcohol dehydrogenase.
- Richard W. Jackson (1930). “A synthesis of tryptophol” (PDF). Journal of Biological Chemistry. 88 (3): 659–662.
- Dickinson, JR; Salgado, LE; Hewlins, MJ (2003). “The catabolism of amino acids to long chain and complex alcohols in Saccharomyces cerevisiae”. The Journal of Biological Chemistry. 278 (10): 8028–34. doi:10.1074/jbc.M211914200. PMID 12499363.
- Pathway: tryptophan degradation VIII (to tryptophol) at BioCyc.org
It is formed from tryptophan, along with indole-3-acetic acid in rats infected by Trypanosoma brucei gambiense.
- Stibbs, H. H.; Seed, J. R. (1975). “Short-Term Metabolism of \14C]Tryptophan in Rats Infected with Trypanosoma brucei gambiense”. Journal of Infectious Diseases. 131 (4): 459–62. doi:10.1093/infdis/131.4.459. PMID 1117200.
An efficient conversion of tryptophan to indole-3-acetic acid and/or tryptophol can be achieved by some species of fungi in the genus Rhizoctonia.
- Toshiko Furukawa, Jinichiro Koga, Takashi Adachi, Kunihei Kishi and Kunihiko Syono (1996). “Efficient Conversion of L-Tryptophan to Indole-3-Acetic Acid and/or Tryptophol by Some Species of Rhizoctonia”. Plant Cell Physiol. 37 (7): 899–905. doi:10.1093/oxfordjournals.pcp.a029037.
Biodegradation
In Cucumis sativus (cucumber), the enzymes indole-3-acetaldehyde reductase (NADH) and indole-3-acetaldehyde reductase (NADPH) use tryptophol to form (indol-3-yl)acetaldehyde.
- Brown HM, Purves WK (1976). “Isolation and characterization of indole-3-acetaldehyde reductases from Cucumis sativus“. J. Biol. Chem. 251 (4): 907–13. doi:10.1016/S0021-9258(17)33779-1. PMID 2607.
Glycosides
The unicellular alga Euglena gracilis converts exogenous tryptophol to two major metabolites: tryptophol galactoside and an unknown compound (a tryptophol ester), and to minor amounts of indole-3-acetic acid, tryptophol acetate, and tryptophol glucoside.
- Laćan, G; Magnus, V; Jericević, B; Kunst, L; Iskrić, S (1984). “Formation of Tryptophol Galactoside and an Unknown Tryptophol Ester in Euglena gracilis”. Plant Physiology. 76 (4): 889–93. doi:10.1104/pp.76.4.889. PMC 1064400. PMID 16663965.
Biological effects of tryptophol and its derivatives
Tryptophol and its derivatives 5-hydroxytryptophol and 5-methoxytryptophol, induce sleep in mice. It induces a sleep-like state that lasts less than an hour at the 250 mg/kg dose. These compounds may play a role in physiological sleep mechanisms. It may be a functional analog of serotonin or melatonin, compounds involved in sleep regulation.
- Cornford, Eain M.; Crane, Paul D.; Braun, Leon D.; Bocash, William D.; Nyerges, Anthony M.; Oldendorf, William H. (1981). “Reduction in Brain Glucose Utilization Rate after Tryptophol (3-Indole Ethanol) Treatment”. Journal of Neurochemistry. 36 (5): 1758–65. doi:10.1111/j.1471-4159.1981.tb00428.x. PMID 7241135. S2CID 9169087.
- Feldstein, A.; Chang, F.H.; Kucharski, J.M. (1970). “Tryptophol, 5-hydroxytryptophol and 5-methoxytryptophol induced sleep in mice”. Life Sciences. 9 (6): 323–9. doi:10.1016/0024-3205(70)90220-1. PMID 5444013.
Tryptophol shows genotoxicity in vitro.
- Kosalec, Ivan; Ramić, Snježana; Jelić, Dubravko; Antolović, Roberto; Pepeljnjak, Stjepan; Kopjar, Nevenka (2011). “Assessment of Tryptophol Genotoxicity in Four Cell Lines in Vitro: A Pilot Study with Alkaline Comet Assay”. Archives of Industrial Hygiene and Toxicology. 62 (1): 41–49. doi:10.2478/10004-1254-62-2011-2090. PMID 21421532.
Tryptophol is a quorum sensing molecule for the yeast Saccharomyces cerevisiae. It is also found in the bloodstream of patients with chronic trypanosomiasis. For that reason, it may be a quorum sensing molecule for the trypanosome parasite.
- Kosalec, Ivan; Ramić, Snježana; Jelić, Dubravko; Antolović, Roberto; Pepeljnjak, Stjepan; Kopjar, Nevenka (2011). “Assessment of Tryptophol Genotoxicity in Four Cell Lines in Vitro: A Pilot Study with Alkaline Comet Assay”. Archives of Industrial Hygiene and Toxicology. 62 (1): 41–49. doi:10.2478/10004-1254-62-2011-2090. PMID 21421532.
- Wuster, Arthur; Babu, M. Madan (2010). “Transcriptional control of the quorum sensing response in yeast”. Molecular BioSystems. 6 (1): 134–41. doi:10.1039/B913579K. PMID 20024075.
In the case of trypanosome infection, tryptophol decreases the immune response of the host.
- Ackerman, S. B.; Seed, J. R. (1976). “The effects of tryptophol on immune responses and its implications toward trypanosome-induced immunosuppression”. Experientia. 32 (5): 645–7. doi:10.1007/BF01990212. PMID 776647. S2CID 12695689.
As it is formed in the liver after ethanol ingestion or disulfiram treatment, it is also associated with the study of alcoholism. Pyrazole and ethanol have been shown to inhibit the conversion of exogenous tryptophol to indole-3-acetic acid and to potentiate the sleep-inducing hypothermic effects of tryptophol in mice.
- Cornford, E. M.; Bocash, W. D.; Braun, L. D.; Crane, P. D.; Oldendorf, W. H.; MacInnis, A. J. (1979). “Rapid distribution of tryptophol (3-indole ethanol) to the brain and other tissues”. Journal of Clinical Investigation. 63 (6): 1241–1248. doi:10.1172/JCI109419. PMC 372073. PMID 447842.
- Cornford, Eain M.; Crane, Paul D.; Braun, Leon D.; Bocash, William D.; Nyerges, Anthony M.; Oldendorf, William H. (1981). “Reduction in Brain Glucose Utilization Rate after Tryptophol (3-Indole Ethanol) Treatment”. Journal of Neurochemistry. 36 (5): 1758–65. doi:10.1111/j.1471-4159.1981.tb00428.x. PMID 7241135. S2CID 9169087.
- Seed, John Richard; Sechelski, John (1977). “Tryptophol levels in mice injected with pharmacological doses of tryptophol, and the effect of pyrazole and ethanol on these levels”. Life Sciences. 21 (11): 1603–10. doi:10.1016/0024-3205(77)90237-5. PMID 600013.
It is a growth promoter of cucumber hypocotyl segments. The auxinic action in terms of embryo formation is even better for tryptophol arabinoside on Cucurbita pepo hypocotyl fragments.
- Rayle, DL; Purves, WK (1967). “Isolation and Identification of Indole-3-Ethanol (Tryptophol) from Cucumber Seedlings”. Plant Physiology. 42 (4): 520–524. doi:10.1104/pp.42.4.520. PMC 1086576. PMID 16656532.
- Jelaska, Sibila; Magnus, Volker; Seretin, Mira; Lacan, Goran (1985). “Induction of embryogenic callus in Cucurbita pepo hypocotyl explants by indole-3-ethanol and its sugar conjugates”. Physiologia Plantarum. 64 (2): 237–242. doi:10.1111/j.1399-3054.1985.tb02342.x.
See also
- Alloisoleucine, the diasteromer of isoleucine
- Wine chemistry
- Indole-3-carbinol
- Indole-3-butyric acid
- Alcohol intolerance
- Disulfiram-like drug
- Mushroom poisoning
- Auxins
- Human drug metabolites
- Hypnotics
- Indoles
- Primary alcohols
- Adenosine receptor antagonists
- Beta1-adrenergic agonists
- Bitter compounds
- Drugs developed by Merck
- Bronchodilators
- Phosphodiesterase inhibitors
- Xanthines
- Histone Acetyltransferase Inhibitor
- Thiols
- Peptides
- NMDA receptor agonists
- Secondary amines
- Polyamines
- Plant hormones
- Alpha-Amino acids
- Antiprotozoal agents
- Hair removal
- Orphan drugs
- Drugs developed by AbbVie
- Fluorinated amino acids
- Difluoromethyl compounds
- Covalent inhibitors
- Drugs developed by Bayer
- Anilines
- Antiprotozoal agents
- Aromatic amines
- Arsenic heterocycles
- Health in Africa
- Organoarsenic dithiolates
- Primary alcohols
- Triazines
- World Health Organization essential medicines
- Health in Africa
- Insect-borne diseases
- Parasitic diseases
- Protozoal diseases
- Sleep disorders
- Tropical diseases
- Zoonoses
- African trypanosomiasis
- Acetaldehyde dehydrogenase inhibitors
- Alcohol abuse
- Alcohol and health
- Antiprotozoal agents
- Danish inventions
- Dopamine beta hydroxylase inhibitors
- Thiuram disulfides
- Disulfiram-like drugs
- COVID-19 drug development
- Substance-related disorders
Leave a Reply